
Die Akromegalie ist eine seltene endokrinologische Erkrankung, unter der in Deutschland ca. 8 000 bis 10 000 Menschen leiden. Aufgrund der schleichenden Symptomatik dauert es ca. acht bis zehn Jahre, bevor die Diagnose gestellt wird. Das ist tragisch, denn in dieser Zeit können sich Komplikationen auch an inneren Organen entwickeln, die das Leben der Patienten deutlich verkürzen. Dieser Artikel soll dazu dienen, frühe Symptome einer Akromegalie rechtzeitig zu erkennen.
Die Akromegalie wird in den meisten Fällen durch ein Wachstumshormon- (GH: growth hormone) produzierendes Hypophysenadenom hervorgerufen. Die meisten Adenome sind Makroadenome (> 1 cm). In weniger als 1 % der Fälle kann auch eine ektope Wachstumshormon- oder GHRH-Produktion wie z. B. bei Bronchialkarzinomen eine Akromegalie hervorrufen [1]. GHRH ist üblicherweise ein aus dem Hypothalamus sezerniertes Releasing-Hormon, welches in der Hypophyse die Wachstumshormon-Produktion stimuliert.
Das Manifestationsalter der Akromegalie liegt zwischen dem 45. und 55. Lebensjahr. Tritt in den sehr seltenen Fällen eine vermehrte Wachstumshormonproduktion vor Schluss der Epiphysen in der Pubertät auf, so entsteht das typische Bild eines Gigantismus. Die längsten Menschen der Welt sind nahezu ausschließlich akromegale Patienten. Einer der bekanntesten Giganten ist Richard Kiel, bekannt durch seine Rolle als „Beißer“ in den James-Bond-Filmen.
Akromegalie am Händedruck erkennen
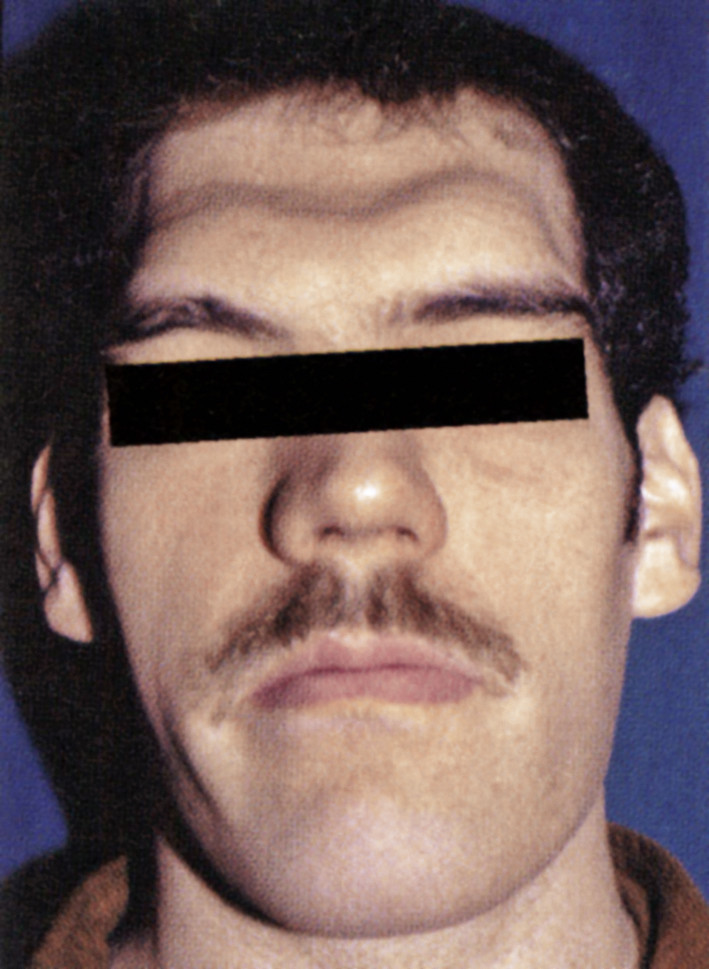
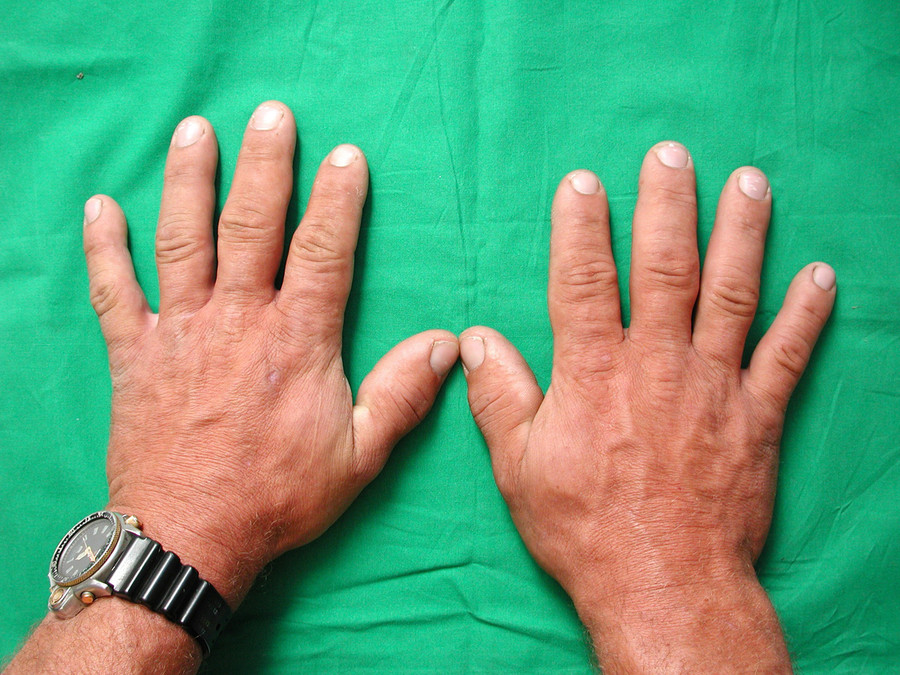
Zu den typischen Symptomen der Akromegalie zählen vergröberte Gesichtszüge mit vergrößerter Nase und prägnanten Wangenknochen (Abb. 1), größer werdende Hände (Abb. 2) und Füße, die durch eine Akkumulation von Mukopolysacchariden im subkutanen Fettgewebe mit folglicher Wasseransammlung entstehen. Darüber hinaus wird eine nahezu permanente Hyperhidrosis (Schweißneigung) angegeben. Somit kann man die Akromegalie bereits beim Händedruck einer großen weichen schwitzigen Hand erkennen.
Lokale Komplikationen durch das wachsende Makroadenom sind Kopfschmerzen sowie Gesichtsfelddefekte durch Komprimierung des Chiasma opticum. Zudem kommen häufiger Gelenkbeschwerden sowie ein Karpaltunnel-Syndrom vor.
Systemische Komplikationen
Durch die Wachstumshormon-induzierte Insulinresistenz kann ein sogenannter Typ-3-Diabetes ähnlich wie beim Typ-2-Diabetiker entstehen. Durch eine vermehrte Natriumrückresorption entsteht eine Hypertonie.
Unbehandelt oder schlecht medikamentös eingestellt können sich weitere systemische Komplikationen entwickeln, unter denen die kardiovaskulären Folgen mit am wichtigsten sind [2, 3]. Der Wachstumshormon- und IGF-1-Exzess führt zu einer Kardiomegalie mit diastolischen und systolischen Funktionsstörungen [4]. Es können außerdem eine Herzinsuffizienz, Klappen-Vitien, Herzrhythmusstörungen und eine koronare Herzerkrankung entstehen [5 – 7].
Auch Malignome können sich ausbilden, so dass insbesondere bei älteren Akromegalen eine Koloskopie zum Ausschluss eines Kolonkarzinoms durchgeführt werden muss. Ebenso werden gehäuft Prostatakarzinome und Schilddrüsenkarzinome (papilläre) beobachtet [8].
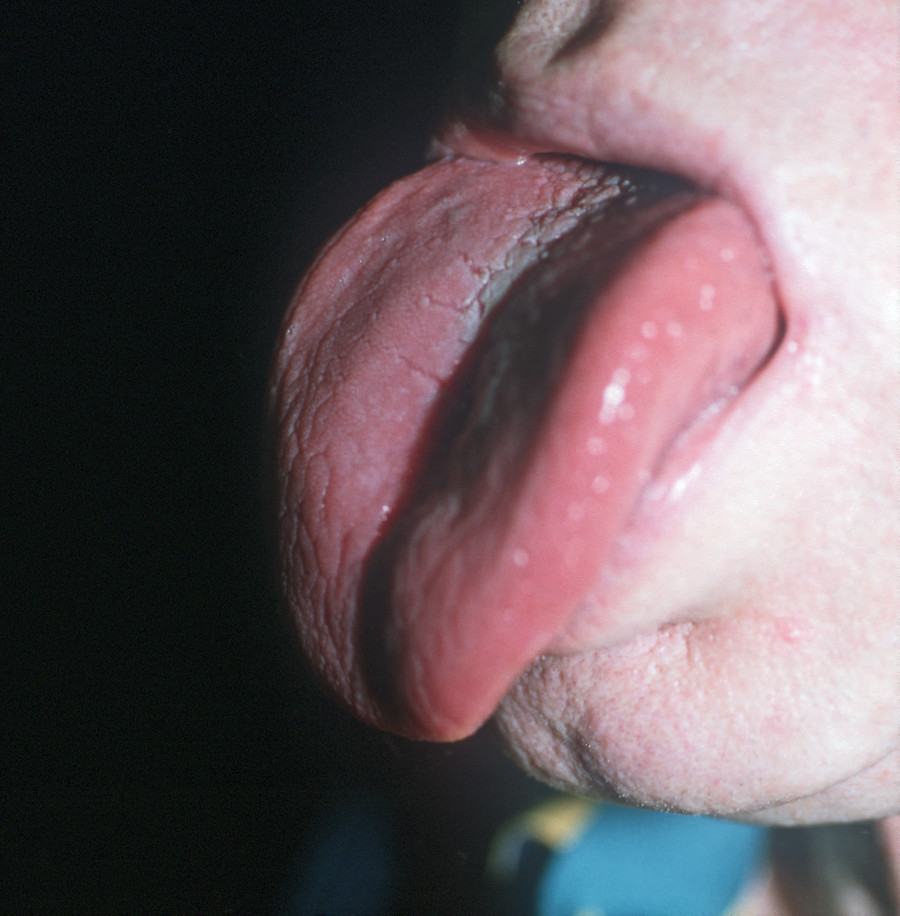
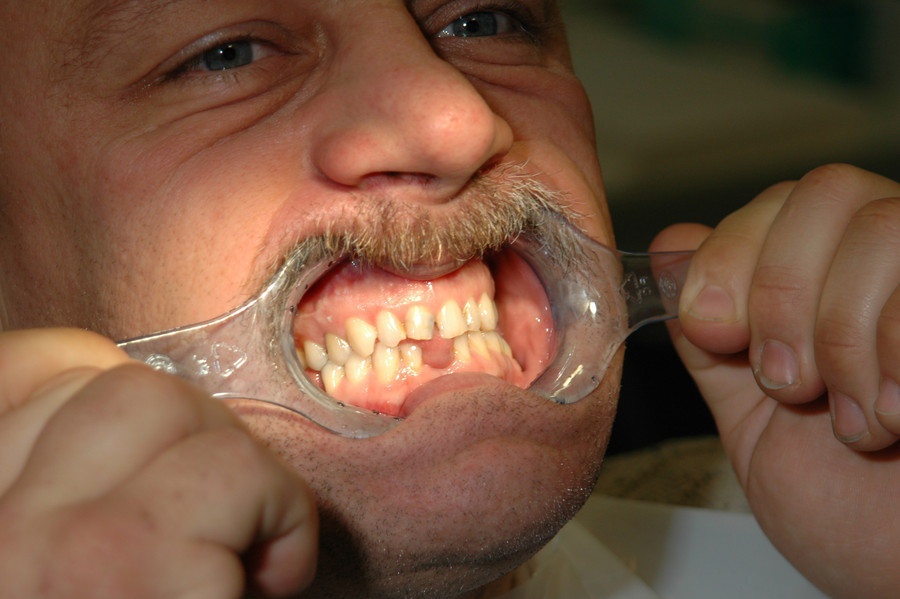
Die Weichteilschwellung betrifft auch die Zunge (Abb. 3), so dass es zu einer Makroglossie kommt. Die Patienten haben deshalb eine leicht verwaschene Sprache, Schluckstörungen und eine deutliche Schnarchneigung wie bei einem obstruktiv bedingten Schlafapnoe-Syndrom. Ca. 30 – 50 % aller Akromegalen leiden unter einem Schlafapnoe-Syndrom [9]. Durch das Kieferwachstum kann es zu einem Zahnausfall kommen. Weitere Zahnveränderungen wie z. B. ein Diastema – eine Lücke zwischen den mittleren Schneidezähnen – oder eine Progenie sind häufig (Abb. 4). Je früher die Erkrankung festgestellt wird, desto wahrscheinlicher ist es, dass diese systemischen Folgeerkrankungen vermieden werden können [10].
Diagnose
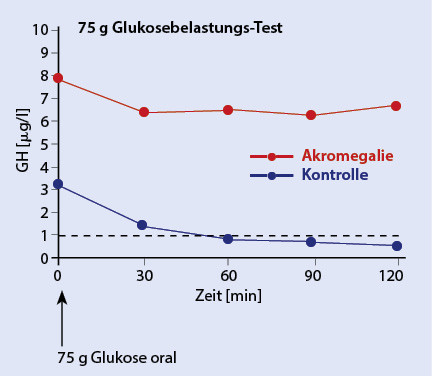
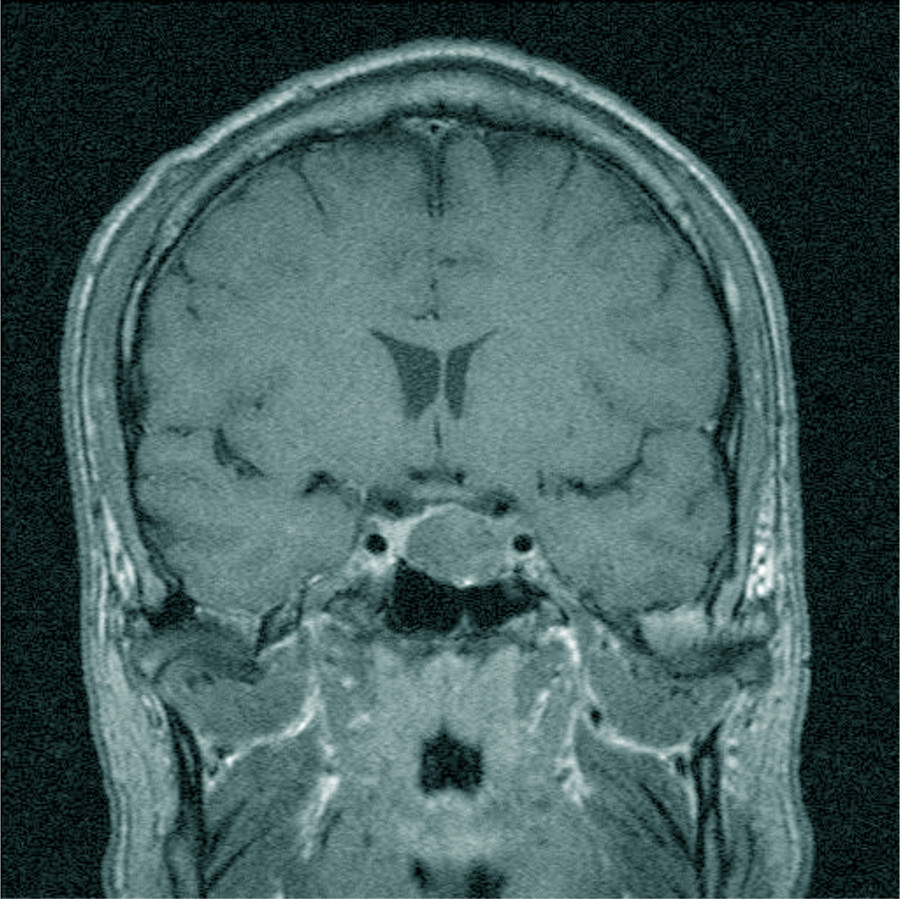
Bei klinischem Verdacht einer Akromegalie sollte durch eine Serumprobe das Wachstumshormon (GH: Growth Hormon) als auch das vorwiegend in der Leber hierdurch synthetisierte IGF-1 (insulin-like-growth-factor-1), früher Somatomedin C genannt, bestimmt werden [11]. Bei erhöhtem IGF-1 muss anschließend ein oraler Glukosetoleranztest mit Bestimmung des Wachstumshormons alle 30 Minuten über zwei Stunden erfolgen (Abb. 5). Bei einer fehlenden Suppression unter 1 ng/ml wird dann die Diagnose der Akromegalie gestellt [12]. Erst danach erfolgt die Bildgebung mittels koronarer Schichtung durch eine MRT-Untersuchung (Abb. 6).
Therapie
Primär sollte bei einer Akromegalie die auf transsphenoidalem Wege durchgeführte Resektion des Hypophysenadenoms angestrebt werden [13]. Diese Methode wurde erstmals durch Harvey Cushing (1869 – 1939) beschrieben. Durch stereotaktische Bestrahlungen können kleine Restadenome bei hinreichendem Abstand zum Chiasma opticum kurativ behandelt werden.
Medikamentös stehen neben den weniger wirksamen Dopaminagonisten wie Bromocriptin oder Cabergolin die deutlich potenteren Somatostatin-Analoga wie Octreotid oder Lanreotid zur Verfügung. Octreotid und Lanreotid können als ein Slow-releasing-Präparat i.m. bzw. tief subkutan alle vier respektive vier bis acht Wochen injiziert werden [14].
Seit einigen Jahren steht auch das wirksamste Medikament zur Hemmung der Wachstumshormonwirkung durch den Wachstumshormon-Rezeptor-Antagonisten Pegvisomant zur Verfügung [15]. Ziel der Therapie ist einerseits, lokale Komplikationen des Makroadenoms wie Sehstörungen oder Kopfschmerzen zu vermeiden. Andererseits müssen systemische Komplikationen mit der daraus resultierenden bis zu zweifach erhöhten Mortalität verhindert werden [16, 17]. Unbehandelt geht die Akromegalie mit einer um acht bis zehn Jahre verkürzten Lebenserwartung einher. Bei adäquater Therapie durch Operation und/oder medikamentöse Therapie ist die Lebenserwartung bei guter Kontrolle nicht reduziert [18].

Erschienen in: Der Allgemeinarzt, 2013; 35 (8) Seite 38-40