

Die sogenannten Orphan Diseases sind dadurch gekennzeichnet, dass jede von ihnen sehr selten vorkommt, d. h. mit einer Häufigkeit von unter 50 Fällen pro 100 000 Einwohnern. Die mangelnde Vertrautheit mit diesen Erkrankungen bringt es mit sich, dass sie oft erst spät diagnostiziert werden. Dabei stehen für einige dieser Waisenkinder wirksame Therapiemöglichkeiten zur Verfügung, die umso besser wirken, je früher sie zum Einsatz kommen. Wir sprachen mit Prof. Dr. med. Thorsten Marquardt, Experte für angeborene Stoffwechselkrankheiten am Universitätsklinikum Münster, über dieses Dilemma.
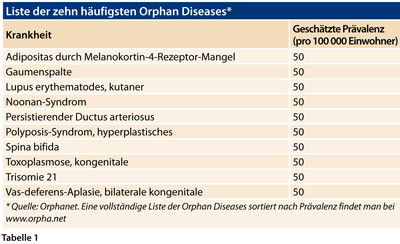
Es stimmt zwar, dass jede der Orphan Diseases für sich genommen sehr selten ist, d. h. definitionsgemäß mit einer Häufigkeit von maximal 1:2 000 (vgl. Tabelle 1) vorkommt. Da es aber mehr als 6 000 verschiedene Orphan Diseases gibt - davon allein an die 1 000 angeborene Stoffwechselerkrankungen - , ist die Gesamtheit der Waisenkinder-Krankheiten durchaus eine respektable Größe, erklärte Prof. Marquardt.
Wegweiser: Ungewöhnliche Symptome
Jeder niedergelassene Kollege wird mindestens einmal während seiner Praxistätigkeit eine dieser seltenen Krankheiten zu sehen bekommen. Die Frage ist nur, ob er sie auch als solche erkennt. Dazu lieferte Prof. Marquardt ein Beispiel: Eine 25-jährige Frau hatte bereits im Alter von drei Monaten Symptome einer Milzvergrößerung entwickelt und Krampfanfälle erlitten - eine ungewöhnliche Kombination von Symptomen. Es hat jedoch 24 Jahre gedauert, bis die zugrunde liegende Krankheit - ein Morbus Gaucher - diagnostiziert wurde. Hellhörig ließ den Hausarzt die milde Anämie werden. Bei der Knochenmarkpunktion fielen dann für die Erkrankung charakteristische Veränderungen auf. Was man braucht, um solche Patienten herauszufischen, ist nicht ein Spezialwissen, so Marquardt, sondern einfach Neugier und Beharrlichkeit, d. h. ein Bestreben, ungewöhnliche Symptome nicht auf sich beruhen zu lassen, sondern weiterzuverfolgen.
Natürlich kann man vom Hausarzt nicht verlangen, die Charakteristika sämtlicher Orphan Diseases ständig parat zu haben. Daher lautet Marquardts Anliegen: Wenn ein ungewöhnliches Symptom auftritt, was man sich nicht ohne Weiteres erklären kann, oder Symptome, die nicht zusammenpassen, dann lohnt es sich, der Sache auf den Grund zu gehen oder den Patienten an einen Spezialisten zu überweisen.
Manifestation von Orphan Diseases
Alle diese angeborenen Stoffwechselerkrankungen können sich bereits - je nach Ausprägung - in der frühen Kindheit äußern oder auch später, manchmal sogar erst im späten Erwachsenenalter. Bei der schweren Form von M. Pompe, einer auf einem Enzymdefekt beruhenden lysosomalen Speicherkrankheit, bei der es zu Funktionsstörungen der Muskulatur kommt, sind z. B. die betroffenen Kinder im Alter von maximal zwölf Monaten tot. Ursache ist die Beteiligung des Herzmuskels, was sich als hypertrophe Kardiomyopathie äußert. Wenn noch eine Restfunktion des Enzyms - der sauren Glukosidase - vorhanden ist, macht sich die Muskelschwäche eventuell erst im Erwachsenenalter bemerkbar.
Eine Reihe dieser lysosomalen Speicherkrankheiten geht mit einer Milzvergrößerung einher. Eine Milzvergrößerung, für die man sonst keine Erklärung hat, sollte also daran denken lassen. Auch Entwicklungsverzögerungen bei Kindern können ein Zeichen für eine solche Stoffwechselerkrankung sein. Allerdings gehen nicht alle Orphan Diseases mit Entwicklungsverzögerungen einher.
Therapiemöglichkeiten
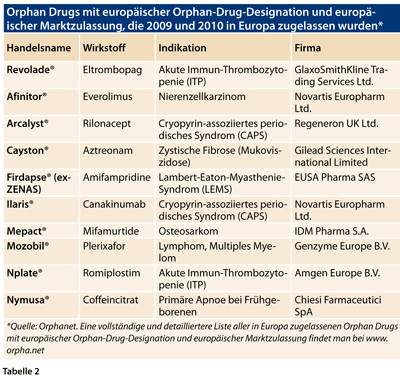
Die Therapiemöglichkeiten angeborener Stoffwechselerkrankungen beruhen darauf, dass man die Enzyme, die die Patienten nicht produzieren können, per Infusion zuführt und damit den fehlenden körpereigenen Stoff ersetzt (vgl. Tabelle 2). Bei M. Pompe und auch bei den Mucopolysaccharidosen stehen solche Enzymtherapien zur Verfügung, die allerdings sehr, sehr teuer sind. Die teuersten Therapien verschlingen bei einem normalgewichtigen Erwachsenen ca. 1 Million Euro pro Jahr. Diese Enzympräparate sind deshalb so teuer, weil es ja nur so wenige Patienten mit diesen Erkrankungen gibt und die Firmen ansonsten ihre Entwicklungs- und Herstellungskosten nicht decken könnten.
Prof. Marquardt sieht die Politik da gefordert: Wenn nur eine begrenzte Geldmenge zur Verfügung steht und die auf alle gerecht verteilt werden soll, muss man sich darüber Gedanken machen, was eine solche Therapie überhaupt kosten darf. Weil die Fallzahlen so klein sind, hatten die Orphan Diseases bisher keinen relevanten prozentualen Anteil am Gesamtgesundheitsbudget. Aber je mehr Krankheiten behandelbar werden, desto mehr wird die Problematik gesundheitspolitisch relevant werden. Und dann wird man vonseiten der Gesundheitspolitik und Gesetzgebung regulierend eingreifen müssen.
Prognose
Mit einer spezifischen Therapie kann man den weiteren Krankheitsverlauf stoppen. Heilen kann man die Erkrankung dagegen nicht. Wenn ein Patient bei Therapiebeginn bereits im Rollstuhl sitzt, bleibt er auch im Rollstuhl sitzen. Wir können die Zeit auch durch eine Enzymtherapie nicht mehr zurückdrehen, so Marquardt.
Wenn man die Erkrankung allerdings schon kurz nach der Geburt entdecken würde, könnte man den Patienten asymptomatisch halten. Dies wäre bei einigen Erkrankungen theoretisch durch ein Neugeborenenscreening möglich, was ja z. B. für die Phenylketonurie bereits durchgeführt wird. Ein Screening würde aber auch nur Sinn machen für die Krankheiten, die man behandeln kann. Abgesehen von den Neugeborenenscreenings ist es in Deutschland nur dann erlaubt, bei einem Kind auf eine angeborene Stoffwechselstörung zu testen, wenn Symptome vorhanden sind und wenn die Krankheit behandelt werden kann. Denn das neue Gendiagnostikgesetz, das seit dem 1.2.2010 gilt, stärkt das Recht auf Nichtwissen. Bei einem Kind, das keine speziellen Symptome oder Befunde hat, die auf eine Orphan Disease hindeuten, darf man also nicht darauf testen, auch wenn die Eltern dies z. B. wegen eines erkrankten Geschwisterkindes wünschen.
Dr. med. Vera Seifert
Erschienen in: Der Allgemeinarzt, 2010; 32 (11) Seite 43-45