
Die C3-Glomerulopathie ist eine äußerst seltene Erkrankung der Niere, deren Inzidenz bei circa 1 bis 2 Fällen pro 1 Million Einwohnern liegt und die zu chronischen glomerulären Schädigungen sowie zum Funktionsverlust der Niere führt. Eine effektive Therapie existiert aktuell nicht. Bei Nierentransplantationen kommt es häufig zu einer Krankheits-Rekurrenz, sodass die Prognose für ein transplantiertes Organ schwierig ist
Die C3-Glomerulopathie repräsentiert eine Gruppe von heterogenen Erscheinungsformen, die in der Regel durch eine defekte Komplementaktivierung ausgelöst werden und die zu einer Schädigung der Glomeruli und der Nieren führen. Somit ist die Modulation oder Inhibition von Komplement eine sinnvolle Therapieoption. Es ist deshalb von großem Interesse, die Mechanismen der defekten Komplement-Regulation und die Pathologie der Erkrankung zu verstehen sowie neue präzise Biomarker zu identifizieren. So lassen sich langfristig gezielte Therapien mit neuen Komplementinhibitoren etablieren, die aktuell in klinischen Studien getestet werden. Damit sind in Zukunft neue Therapieformen für diese schwere Nierenerkrankung zu erwarten.
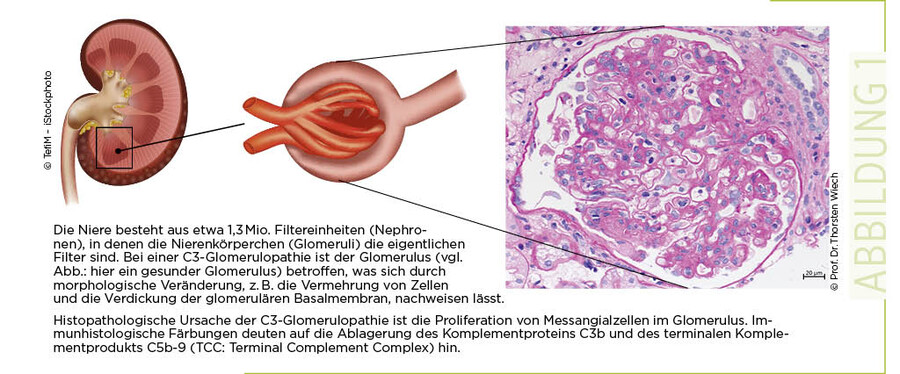
Morphologische und histologische Veränderungen bei C3-Glomerulopathie
Die C3-Glomerulopathie ist eine heterogene Erkrankung, die unterschiedliche morphologische und histologische Änderungen in verschiedenen Segmenten der Niere verursachen kann (1, 2). Eine Nierenbiopsie ist das beste Vorgehen, um diese Erkrankung zu diagnostizieren. Die Biopsie erfasst sowohl morphologische als auch immunhistologische Veränderungen. Morphologische Veränderungen zeigen sich dem Nierenpathologen in Form einer Veränderung, einer Verdickung der glomerulären Basalmembran (intramembranöse Glomerulonephritis mit "Dense Deposit Disease Pattern") sowie einer Vermehrung der mesangialen Zellen, der extrakapillären Zellproliferation und der Proliferation der Zellen im subendothelialen Raum (membranoproliferatives Muster).
Häufig ist eine Infiltration von inflammatorischen Makrophagen zu beobachten (3) (Abb. 1).
Charakteristische immunhistochemische Merkmale der C3-Glomerulopathie sind eine massive Deposition von C3b in der Niere und keine oder ein häufig nur sehr geringer Nachweis von Immunglobulinen (IgGs) (4) (Abb. 1). Weitere Komplementproteine, wie Properdin und der terminale Komplementkomplex (C5b-9) können oft im Glomerulus nachgewiesen werden. Diese Veränderungen sind in der Regel chronischer Natur, die zu fortlaufenden Entzündungsprozessen und zu Gewebeschädigung führen und parallel auch Reparaturprozesse auslösen. Diese chronischen Prozesse können erklären, warum sich eine C3-Glomerulopathie über einen längeren Zeitraum, in der Regel über Jahre, entwickelt und manche Therapeutika keine sofortigen messbaren Effekte in der Niere induzieren (4).
Ursachen und Pathophysiologie
Eine veränderte Komplementfunktion ist eine häufige Ursache der C3-Glomerulopathie. Das Komplement hat als zentrales Immunabwehrsystem des menschlichen Organismus vielfältige Aufgaben (5). Ein wichtiger Aspekt dieses Abwehrsystems ist es, die Homöostase im Organismus aufrechtzuerhalten. Ein aktiviertes Komplement bindet an veränderte körpereigene Zellen wie apoptotische oder nekrotische sowie an infektiöse Erreger und vermittelt so deren direkte und effiziente Beseitigung durch körpereigene Fresszellen. Ebenfalls bildet das aktivierte Komplementsystem immunbiologische Botenstoffe, die Entzündungszellen zum Ort des Immungeschehens locken können.
Da es schneller als alle anderen Teile des Immunsystems reagiert, kann das Komplementsystem als immunbiologischer "Ersthelfer" betrachtet werden, der das weitere Immungeschehen orchestriert und Entzündungsreaktionen steuert.
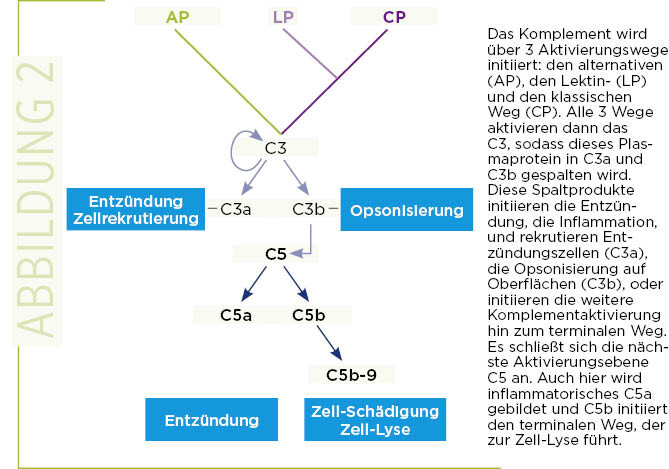
Ein Komplement hat drei Hauptaktivierungswege: den alternativen (AP), den Lektin- (LP) und den klassischen Aktivierungsweg (CP). Jeder Weg wird durch unterschiedliche Proteine initiiert, alle 3 führen zur Neubildung von Enzymen, den Konvertasen. Jeder Aktivierungsweg führt zur Bildung einer C3-Konvertase, die das zentrale Komplementprotein C3 spaltet und die Effektorproteine C3a und C3b generiert.
Im nächsten Schritt werden weitere neue Enzyme gebildet, die in analoger Weise C5 zu C5a und C5b spalten. C3a und insbesondere C5a sind Botenstoffe, die Entzündungszellen an den Ort des Immungeschehens dirigieren und dort Entzündungsreaktionen auslösen. C3b bindet an der Oberfläche von veränderten Körperzellen und fremden Zellen, markiert diese Partikel und steuert so deren Beseitigung. Der dritte zentrale Reaktionsschritt ist die Bildung des terminalen Komplexes (TCC), der auch als C5b-9 oder Membranangriffs-Komplex bezeichnet wird. Das Komplement ist ein hocheffizientes Effektor-System, das aus fast 100 einzelnen Komponenten und Aktivierungsfragmenten besteht. Die einzelnen Bestandteile der Kaskaden, wie C3 und C5, und deren Aktivierungsfragmente zirkulieren in der Regel im Plasma. Aktivatoren induzieren und verstärken die Kaskade, Regulatoren bestimmen den genauen Typ der Reaktion und steuern die Stärke der Aktivierung, während Inhibitoren in der Lage sind, die Kaskade auf jeder einzelnen Ebene zu blockieren. Wie aggressiv und zerstörerisch das Komplementsystem wirken kann, wird zum Beispiel bei Abstoßung von Transplantaten sichtbar, wobei das Organ komplett zerstört werden kann (5).
Ausgehend von dieser komplexen Gegebenheit der Aktivierung und Inhibition ist das Gleichgewicht des Systems von großer Bedeutung. Jegliche Dysfunktion führt zur Pathologie und bei der C3-Glomerulopathie zum Funktionsverlust der Niere. Deshalb muss man verstehen, wo die Kaskade angeschaltet wird: im Blut/Plasma oder direkt auf der Zelloberfläche, welche Schritte der Kaskade aktiviert werden oder auf welcher Ebene sie nicht richtig reguliert wird. So lässt sich besser nachvollziehen, wie es zu Veränderungen und zur Schädigung des Glomerulus der Niere kommt.
Ursachen der C3-Glomerulopathie
Zum Verständnis der Pathogenese der C3-Glomerulopathie muss man bestimmen, ob vermehrt Aktivierungsfragmente gebildet werden, welches Komplementprotein genau oder welcher Regulator modifiziert ist und welche Aktivierungsfragmente den Glomerulus schädigen. Daher ist es neben einer Nierenbiopsie für die Diagnose hilfreich, Komplementparameter im Plasma zu bestimmen (Abb. 2).
Erworbene autoimmune Faktoren sind häufig
Autoantikörper lassen sich bei Patienten mit C3-Glomerulopathie relativ häufig nachweisen. Die Autoantikörper werden unterschieden in C3-, C4- und C5-Nephritischer Faktor.
C3-Nephritischer Faktor (C3-Nef) ist ein Autoantikörper, der an die C3-Konvertase des AP-Aktivierungsweges bindet und der auch den Amplifikationsloop steuert sowie verstärkt; C5-Nephritischer Faktor ist ein Autoantikörper, der an die C5-Konvertase bindet und das Komplement auf der Ebene von C5 dereguliert; während C4-Nephritischer Faktor, der an die C3-Konvertase des klassischen Komplementweges (CP) bindet, diesen Weg aktiviert. Diese Autoantikörper, die an verschiedene Konvertasen binden, stabilisieren die jeweiligen Enzyme, verlängern deren biologische Halbwertszeit und verstärken so die Komplementaktivierung der einzelnen Aktivierungswege.
Weitere Autoantikörper sind bei C3-Glomerulopathie-Patienten nachgewiesen worden. Diese Antikörper binden den zentralen Inhibitor Faktor H oder Faktor B, die Spaltprodukte C3b, iC3b sowie C3c (6). Eine Quantifizierung der einzelnen Autoantikörper ist von großem Interesse, um den Verlauf der Autoantikörper über die Zeit zu bestimmen und um den Autoantikörper-Spiegel mit dem Schweregrad der Komplementdefekte zu korrelieren. So lässt sich gezielt der betroffene Komplementweg inhibieren und zeitnah der Effekt einer komplement- oder autoantikörperbasierten Therapie verfolgen.
Genetische Ursachen, Mutationen und chromosomale Umordnung
Mehrere Genmutationen wurden bei C3-Glomerulopathie-Patienten nachgewiesen. Diese betreffen die Faktor-H-Gen-Gruppe, die für die Regulatoren Faktor H, FHR1 bis FHR5 kodiert (7). Weitere Veränderungen zeigen die Gene C3, Faktor B und Faktor I. Diese Änderungen können als Punktmutation auftreten, die zum Austausch einzelner Proteinbausteine und Aminosäuren führen, sowie als Deletionen und Duplikationen auf der Ebene einzelner Gene bzw. chromosomaler Segmente. Bei der Beurteilung der einzelnen genetischen Veränderungen ist es sehr wichtig, zwischen krankheitsverursachenden, pathologischen Mutationen und natürlichen Genvarianten, sog. Gen-Polymorphismen, zu unterscheiden. Eine klare Unterscheidung und Zuordnung ist auch für Experten nicht immer einfach (7,8).
Diagnostik der C3-Glomerulopathie
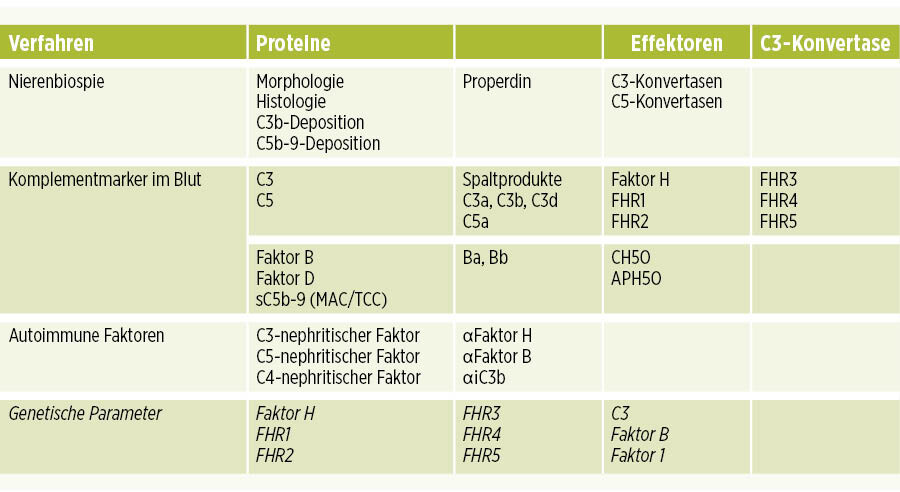
Da eine C3-Glomerulopathie durch ein defektes Komplement verursacht wird, hat sie verschiedene Ursachen (Abb. 2). Deshalb ist neben der Diagnose mittels Nierenbiopsie eine Analytik des Komplementsystems im Blut/Plasma oder im Urin von Bedeutung. Dies beinhaltet den Nachweis der neu entstandenen Spaltprodukte als Marker für eine Aktivierung der jeweiligen Kaskade. Standardtests bestimmen die Komplementaktivierung mittels APH50, CH50, zudem gibt es weitere Komplementaktivierungs-Assays (Tabelle I).
Der Nachweis und die Plasmaspiegel von einzelnen Proteinen – C3, C4 und C5 – sowie der Regulatoren –Faktor H, FHR1, FHR2, FHR3, FHR4, FHR5, Faktor B, Faktor D – sind für die Diagnostik und für die Interpretation der Komplementaktivierung bedeutsam. Darüber hinaus sind der Nachweis und die Quantität von C3-Spaltprodukten – als C3b, iC3b und C3c – sowie der Spaltprodukte von Faktor B (Ba, Bb), von C5 (C5a), und des löslichen terminalen KomplexessTCC (oder sC5b-9) informativ. Anhand des Gehalts dieser Proteine und des Vorhandenseins der Menge einzelner Spaltprodukte kann der Grad der Aktivierung bestimmt und verfolgt werden (Tabelle 1).
Eine einheitliche und zentrale Diagnostik ist für Patienten mit Verdacht auf C3-Glomerulopathie vorteilhaft. Ausgehend davon, dass die möglichen veränderten Komplementparameter nicht großflächig von allen Laboratorien angeboten werden, ist es von großem Interesse, diese Testung zu verbreiten, um bei allen Patienten mit Verdacht auf C3-Glomerulopathie diese wichtigen Parameter zu bestimmen. So lassen sich neben einer präzisen Diagnose auch Therapieerfolge mittels Komplementinhibitoren zeitnah verfolgen. Allerdings sind diese Analysen nicht Gegenstand aktueller Routineuntersuchungen, sondern werden in der Regel nur von Speziallaboren angeboten.
Verstärkte Komplementaktivierung bei der C3-Glomerulopathie
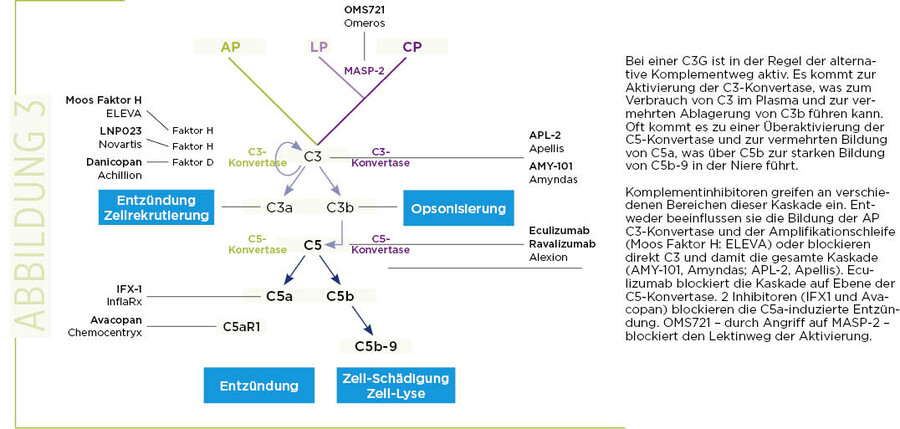
Die Integration der einzelnen Komplementparameter, die bei vielen C3-Glomerulopathie-Patienten erhöht bzw. verändert sind – wie in der "Komplementlandkarte" angezeigt – führt dazu, dass oft eine verstärkte Aktivierung des alternativen Wegs vorliegt, was sich durch vermehrte C3b und C5a darstellen lässt.
Oft lässt sich auch eine Zunahme des terminalen Komplementkomplexes beobachten, der im Plasma als lösliches sC5b-9 und in der Nierenbiopsie als abgelagerte C5b-9 nachgewiesen wird (Abb. 3).
Das heterogene Krankheitsbild der C3-Glomerulopathie mit den verschiedenen Unterformen zeigt sich auch an heterogenen morphologischen und immunhistologischen Erscheinungsformen, einem unterschiedlichen Profil der Komplementparameter sowie an verschiedenen genetischen, autoimmunen Formen. Die richtige Auswahl und die geeignete Kombination dieser Parameter erlaubt es, die heterogenen Formen zu definieren und Untergruppen, basierend etwa auf autoimmunen, durch Aktivierung bedingte oder genetische Marker, zu beschreiben. Langfristig können so gezielt Patienten ausgewählt werden, die von einer Behandlung mit einem ausgewählten Komplementinhibitor profitieren können. Daher wird sich die C3-Glomerulopathie vermutlich als ein gutes Beispiel für eine personalisierte Therapie einer schweren Nierenerkrankung entwickeln.
Komplement-Therapeutika
Die gezielte Entwicklung von Komplementinhibitoren hat zu neuen Komplement-Therapeutika geführt, die sich aktuell in der klinischen Testung befinden.
Die einzelnen Inhibitoren unterscheiden sich sowohl in ihrem Angriffspunkt in der Kaskade als auch in ihrer Art: Diese Inhibitoren sind zum einen rekombinant hergestellte endogene Regulatoren, adaptierte, synthetische monoklonale Antikörper sowie kleinere Peptide. Entsprechend der Angriffspunkte in der "Komplement-Landkarte" lassen sich die für C3-Glomerulopathie ausgewählte Wirkstoffe aufteilen (8, 9).
- Inhibition der C3-Konvertase des alternativen Komplementwegs. Mehrere Wirkstoffe agieren auf Ebene der C3-Konvertase des alternativen Komplementwegs. Dazu gehören der rekombinante, in Moos exprimierte Faktor H (ELEVA), kleinere Peptidwirkstoffe, die an Faktor B (LPN023; Novartis) oder Faktor D (ACH44701; Achillion) binden und die diese Serin-Proteasen inhibieren (Abb. 3).
- Blockierung des Lektinwegs durch einen MASP-2-spezifischen mAB. Der Lektinweg ist für die Aktivierung des Komplements von großer Bedeutung und entsprechend werden Ansätze zur Blockade dieses Aktivierungswegs durch gerichtete Inhibition von MASP-2 (MBL-assoziierte Serinprotease 2) verfolgt.
- Blockade auf Ebene von C3. Peptide, welche direkt an C3 binden und welche die Komplementaktivierung blockieren, sind die Wirkstoffe APL-2 (Apellis) und AMY-101 (Amyndas). Beide haben einen ähnlichen Angriffspunkt wie diejenigen, welche die C3-Konvertase des AP-Wegs inhibieren (Abb. 3). Diese Peptide blockieren jedoch alle 3 Aktivierungswege und haben somit einen stärkeren Effekt auf die Komplementaktivierung als die selektiv auf den AP gerichteten Wirkstoffe.
- Die Blockierung von Komplement auf Ebene der C5-Konvertasen. Ein monoklonaler Antikörper, der an C5 bindet –Eculizumab –, und eine modifizierte Variante mit längerer biologischer Verfügbarkeit (Ravulizimab, beide Alexion) sind bereits in der Klinik zugelassene Medikamente. Eculizumab wird für die Therapie des atypischen hämolytisch-urämischen Syndroms,der paroxysmalen nocturnalen Hämoglobinurie sowie bei 2 weiteren Erkrankungen eingesetzt. Eculizmab wurde bereits bei C3-Glomerulopathie-Patienten verwendet, und die Ergebnisse waren unterschiedlich. Bei einigen, aber nicht bei allen Patienten zeigte der Wirkstoff einen positiven Effekt.
- Blockade der komplementvermittelten Inflammation. Eine weitere Gruppe von Wirkstoffen blockiert selektiv die komplementvermittelte Inflammation auf der C5a- und C5aR1-Achse. IFX1 (InflaRx) ist ein monoklonaler Antikörper, der an C5a bindet und die inflammatorische Aktivität von C5a blockiert. Avacopan (Chemocentryx) bindet an den C5aR1-Rezeptor und hat so einen fast identischen Effekt.
Die Mehrheit dieser Wirkstoffe sind in klinischen Untersuchungen für die C3-Glomerulopathie und werden in Phase II – Phase III evaluiert. Darüber hinaus wird ihre Wirksamkeit auch bei weiteren komplementvermittelten Erkrankungen getestet. Basierend auf der Heterogenität und den vielfältigen Ursachen der C3-Glomerulopathie ist zu erwarten, dass mehrere dieser Inhibitoren für eine Therapie der unterschiedlichen Formen verwendet werden.
Schlussfolgerung und Ausblick
Derzeit geht es darum, die Patienten für die jeweiligen Wirkstoffe zu stratifizieren:Mehrere Komplementinhibitoren werden in klinischen Studien getestet, und es ist davon auszugehen, dass in absehbarer Zukunft Therapeutika für diese schwere Nierenerkrankung verfügbar sein werden. Ausgehend von den komplexen Ursachen der C3-Glomerulopathie ist anzunehmen, dass ein einzelner Inhibitor nicht bei der Behandlung von allen C3-Glomerulopathie-Patienten und allen Unterformen erfolgreich sein wird. Vermutlich werden verschiedene Wirkstoffe für die Therapie eingesetzt werden. Deshalb werden eine Ausweitung, eine Intensivierung, eine Präzisierung, eine Standardisierung sowie eine Vereinheitlichung der Diagnostik für die Erkrankungen umso mehr notwendig. Damit lassen sich weitere Biomarker oder eine Kombination von Biomarkern auswählen, die eine Stratifizierung von Patienten ermöglicht.Es sollte die Behandlung entsprechend der Unterform dieser schwierigen Erkrankungen mit einer Auswahl von verschiedenen Inhibitoren realisierbar sein. Dies wird das Ziel künftiger erfolgreicher Therapieoptionen sein müssen.

Erschienen in: Der Nierenarzt, 2020; 7 (16) Seite 8-13