


Erwachsene mit angeborenen Herzfehlern (EmaH) sind in der Allgemeinarztpraxis längst keine Seltenheit mehr – ihre Zahl steigt kontinuierlich an. Da EmaH auch nach chirurgischer oder interventioneller Behandlung chronisch herzkrank sind, hat der Hausarzt hier eine wichtige Aufgabe: potenzielle Komplikationen bei angeborenem Herzfehler (AHF) erkennen, diese richtig behandeln (lassen) und den Patienten in die so notwendige lebenslange Nachsorge führen. Das Überleben, der Krankheitsverlauf und die Lebensqualität der Betroffenen hängen davon ab.
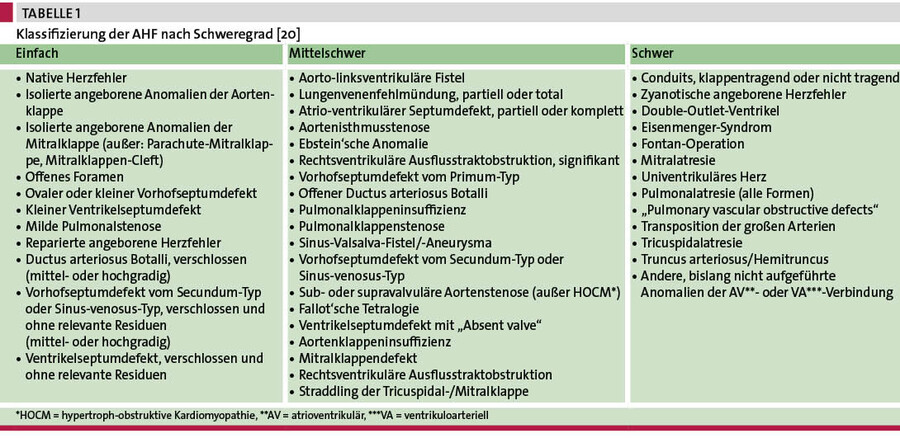
AHF zählen zu den häufigsten isolierten Organanomalien. Bis Ende der 1930er-Jahre verstarben circa 80 % der Kinder mit einem relevanten AHF in den ersten Lebensjahren. Heute erreichen die meisten Patienten mit AHF das Erwachsenenalter und ihre Zahl steigt aufgrund der immer besseren Prognose stetig an. Die Zahl von EmaH liegt bei mehr als 300.000 in Deutschland [11]. Das Spektrum umfasst überwiegend operativ oder interventionell behandelte sowie teilkorrigierte oder native, unbehandelte AHF. Tabelle 1 zeigt eine Einteilung der AHF nach Schweregrad [20]. Komplexe Formen sind selten unbehandelt, werden aber im Rahmen der aktuellen Migrationsbewegungen immer häufiger beobachtet [15].
Trotz aller Behandlungsfortschritte sind AHF nicht vollständig heilbar und gehen fast immer mit Rest- und Folgezuständen oder einem erhöhten Risiko einher, etwa für Herzinsuffizienz und -rhythmusstörungen, pulmonale Hypertonie, Aortopathien oder Endokarditis [17, 21]. Mit Restzuständen sind anatomische oder hämodynamische Normabweichungen gemeint, die Teil der angeborenen Fehlbildung sind und auch nach der Operation bestehen bleiben. Unter Folgezuständen versteht man anatomische oder hämodynamische Nachwirkungen, die aus einem spezifischen Eingriff herrühren, sich als Folge des AHF entwickeln und bei Eingriff nicht vermeidbar waren. Diese Residualzustände sind für jeden AHF und therapeutischen Eingriff spezifisch und für den behandelnden Arzt besonders relevant. Er muss sie, auch als Hausarzt, kennen, um den Patienten adäquat führen und beraten zu können. Mit zunehmendem Alter können sekundäre Komorbiditäten, wie arterielle Hypertonie, koronare Herzkrankheit, Stoffwechselstörungen, zerebrovaskuläre Erkrankungen oder andere Organschäden den Krankheitsverlauf negativ beeinflussen [15]. Die Betroffenen müssen daher nicht selten mit einem Leben, das von permanenter Unsicherheit und Angst geprägt ist, klar kommen. Aktuell leidet die Hälfte aller EmaH (48 %) an psychischen Erkrankungen [2, 24].
Herzinsuffizienz
Ein Großteil der Patienten mit AHF entwickelt im Langzeitverlauf eine Herzinsuffizienz. Dies gilt besonders für Fälle, in denen der morphologisch rechte Ventrikel in die Aorta pumpt [8]. Hinzu kommen Patienten mit nur einer Herzkammer, die häufig eine sogenannte Fontan-Operation erhalten haben, sowie mit schwerer pulmonalvaskulärer Erkrankung (Eisenmenger-Syndrom) oder mit schwerwiegenden Klappenerkrankungen (z.B. nach Fallot-Korrektur) [21].
Eine Herzinsuffizienz beeinflusst die Mortalität und Morbidität von EmaH erheblich und ist bei circa 25 % der Patienten für eine vorzeitige Sterblichkeit verantwortlich [5]. Das Erkennen einer Herzinsuffizienz ist oft nicht einfach, da sich diese oft anders als bei erworbenen Herzerkrankungen manifestiert. Die Betroffenen selbst bemerken eine Herzinsuffizienz erst spät, da sich eine hierfür typische Leistungseinschränkung oft schleichend entwickelt [15]. Die Behandlung der Insuffizienz bei EmaH erfordert spezifische Fachkenntnisse über herzfehlertypische Besonderheiten, da sie von der üblichen Therapie bei erworbenen Herzerkrankungen abweichen kann [17].
Herzrhythmusstörungen
Supraventrikuläre und ventrikuläre Tachykardien sowie bradykarde Herzrhythmusstörungen zählen zu den häufigsten Todesursachen bei EmaH [19]. Dies gilt besonders für Patienten mit Vorhofseptumdefekt, Fallot’scher Tetralogie, Ebstein’scher Anomalie sowie bei komplexeren AHF nach Vorhofumkehr- oder Fontan-Operation [15]. Die Behandlung richtet sich prinzipiell nach dem Vorgehen bei kardiologischen Patienten und kann pharmakologisch, katheterinterventionell, durch Schrittmacher- oder Defibrillatorimplantation erfolgen. Dabei sind vitienspezifische Besonderheiten zu beachten, da diese Eingriffe teils sehr kompliziert sind und sogar eine eigene Leitlinie mit detaillierten Empfehlungen zur Arrhythmiebehandlung bei EmaH notwendig machten [12, 23].
Pulmonale und pulmonal-arterielle Hypertonie
Die pulmonale/pulmonal-arterielle Hypertonie (P(A)H) ist eine schwere Komplikation, die bei vielen AHF und in allen Altersstufen – vom Säugling bis zum Greis – auftreten kann. Da sich die P(A)H bei AHF gravierend auf Belastbarkeit, Lebensqualität und -dauer auswirkt [9],
ist eine frühe, korrekte Diagnose essenziell, um den Verlauf interventionell, operativ oder medikamentös günstig zu beeinflussen. Da sich P(A)H bei AHF oft grundlegend von anderen PAH-Formen unterscheidet, ist eine enge Zusammenarbeit mit EmaH-Spezialisten nötig, um bei P(A)H-spezifischer Therapie bis hin zu intensivmedizinischen Maßnahmen, Operationen oder Lungen-/Herz-Lungen-Transplantation richtig zu entscheiden [4, 6, 9].
Infektiöse Endokarditis
EmaH haben ein besonders hohes Risiko für eine infektiöse Endokarditis (IE), vor allem bei komplexen AHF, bei zyanotischen Patienten oder nach Herzklappenersatz [14]. Trotz moderner Antibiotika und fortgeschrittener, frühzeitiger herzchirurgischer Behandlung ist die Letalität einer IE immer noch hoch, besonders bei später Diagnosestellung. Der Arzt sollte daher gerade bei EmaH stets an die gefährliche Komplikation
der IE denken. Obwohl die aktuelle Leitlinie der European Society of Cardiology eine Endokarditisprophylaxe nur noch für Hochrisikopatienten empfiehlt, sehen einige Spezialisten für EmaH durchaus ein breiteres Indikationsspektrum als für andere kardiologische Patienten [1, 8]
Psychische Störungen
Während das Forschungsinteresse bislang vorwiegend der Diagnostik und der Behandlung von AHF galt, sind die psychischen Auswirkungen der Erkrankung noch unzureichend untersucht. Obwohl EmaH eine gute allgemeine Lebensqualität aufweisen, ist die gesundheitsbezogene Lebensqualität im Vergleich zur Allgemeinbevölkerung deutlich reduziert [3, 13]. Die aktuelle Studienlage deutet zudem darauf hin, dass EmaH ein erhöhtes Risiko für psychische Komorbiditäten wie Angst, Depression und posttraumatische Belastungsstörungen haben [2]. Sie benötigen deshalb eine lebenslange, integrative Nachsorge, die weit über die rein medizinische Behandlung hinausgeht.
EmaH-Versorgung in Deutschland
Für die Versorgung der in Deutschland lebenden EmaH wurde vor einigen Jahren, unter Federführung verschiedener Fachgesellschaften, Fachverbände und Patienteninitiativen, ein dreistufiges Versorgungssystem zur interdisziplinären Versorgung erarbeitet (Abb. 1).
Das Ziel: eine flächendeckende, bedarfsgerechte Versorgung, um Patienten mit AHF – in Abhängigkeit von Art, Schweregrad und Stadium ihres Herzfehlers – lebenslang spezifisch betreuen zu können. In der ersten Versorgungsstufe, der Basisversorgung, werden alle Patienten grundsätzlich durch ihren Hausarzt, Allgemeinmediziner oder hausärztlich tätigen Internisten betreut – vielfach müssen diese Ärzte allerdings noch über die Bedürfnisse dieser Patienten aufgeklärt werden. Die Betreuung sollte dabei in enger Kooperation mit der zweiten Versorgungsstufe, den regionalen EmaH-Schwerpunktpraxen/-kliniken, erfolgen. Diese Einrichtungen wiederum stehen in engem Kontakt mit der dritten Versorgungsebene, den überregionalen EmaH-Zentren (Maximalversorger) [16].
Versorgungsproblematik
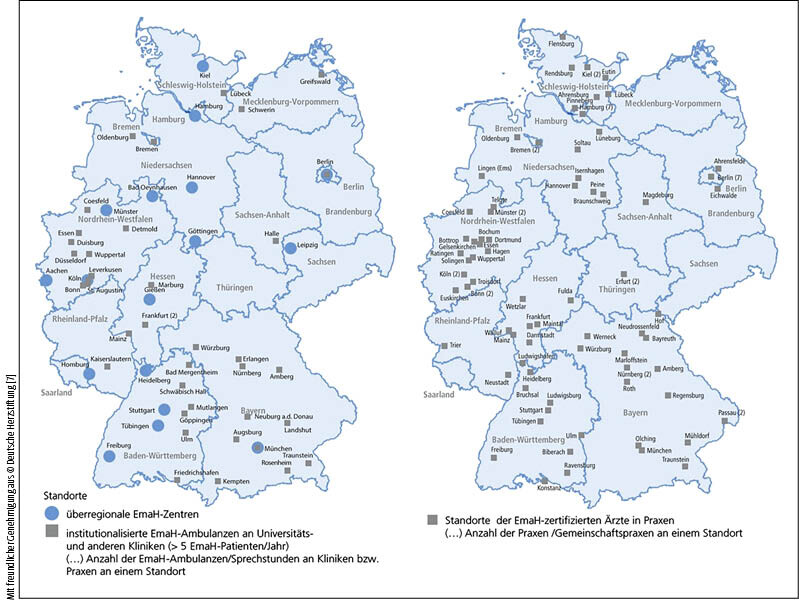
Trotz der flächendeckenden Versorgungsstrukturen für EmaH in Deutschland (vgl. Abb. 2, 3)
gibt es ein Versorgungsdefizit. Da etwa nur ein Drittel der mehr als 300.000 hier lebenden EmaH in zertifizierter Betreuung ist, gelten sie sogar als die am schlechtesten versorgten kardiologischen Patienten [16]. Anders als EmaH sind Kinder und Jugendliche mit AHF bis zum Erreichen der Transitionsphase (Übergang vom Jugend- ins Erwachsenenalter) durch ihre Kinderkardiologen zumeist sehr gut versorgt. Systembedingt müssen die jungen Erwachsenen dann aber diese Versorgungsstrukturen verlassen und neue Ansprechpartner finden. Diese kritische Lebensphase verlangt viel Selbstdisziplin von den heranwachsenden Patienten ab. Dabei unterschätzen sie oft die Bedeutung notwendiger Check-ups und entziehen sich der geregelten Nachsorge, was zu der "Loss to follow-up"-Problematik führt [11]. Diese ist gefährlich, da alle Patienten, unabhängig davon, ob der AHF nativ oder korrigiert ist, chronisch herzkrank sind. Dies gilt nicht nur für komplexe Herzfehler, sondern auch für vermeintlich einfache, die im Langzeitverlauf häufiger als vermutet schwere Komplikationen entwickeln können. Ein Beispiel: die pulmonale Hypertonie bei einfachem Shunt-Vitium, bei der es – trotz frühzeitiger Behandlung – zu erheblichen kardiovaskulären Problemen im Langzeitverlauf kommen kann [11, 22].
Die Rolle des Hausarztes
Aktuelle Daten der VEmaH-Studie ("Versorgungssituation von EmaH") zeigen, dass der Großteil der EmaH bei allen Gesundheitsproblemen (auch den AHF direkt betreffend) als ersten Ansprechpartner ihren Haus-/Allgemeinarzt aufsucht [18]. Die VEmaH-Studienergebnisse verdeutlichen aber leider auch, dass weder die EmaH selbst noch deren Haus-/Allgemeinärzte und hausärztlich tätige Internisten über die spezifische Nachsorge bei AHF ausreichend informiert sind. Weitgehend unbekannt sind dementsprechend die vorhandenen Anlaufstellen. Diese Ergebnisse erschrecken, da Ärzte der Basisversorgung eigentlich "Weichensteller" sein und die Patienten vor allem zur regelmäßigen Vor- und Nachsorge in spezialisierten Einrichtungen anhalten sollten. Gelingt es aber, die Zusammenarbeit zwischen Basisversorgern sowie zertifizierten EmaH-Spezialisten und -Zentren zu verbessern, gibt es gute Chancen, die Morbidität und Mortalität weiter zu senken [18].

Interessenkonflikte: Die Autoren haben keine deklariert
Erschienen in: Der Allgemeinarzt, 2020; 42 (18) Seite 34-40