



Starke Bauchschmerzen, Müdigkeit, lichtempfindliche Haut. Das sind nur einige der möglichen Anzeichen einer Porphyrie, einer Störung der Hämsynthese. Porphyrien umfassen insgesamt acht angeborene, seltene Stoffwechselerkrankungen (mit Ausnahme der erworbenen PCT), die alle eines gemeinsam haben: den Defekt eines Enzyms der Hämbiosynthese. Die jeweilige Enzymdefizienz führt zu einer typischen Akkumulation von Porphyrinvorläufern und/oder Porphyrinen und sehr unterschiedlichen Symptomen.
Häm ist Bestandteil einer Vielzahl von Hämproteinen, wie Hämoglobin, Myoglobin, Cytochrom P450, mitochondrialen Cytochromen, Katalasen und Peroxidasen. Die ubiquitäre Synthese von Häm in allen kernhaltigen Zellen ist für das Leben essenziell. Häm wird überwiegend im Knochenmark (75 – 80 %) und in der Leber (15 – 20 %) in einem mehrstufigen Prozess synthetisiert. Aus Succinyl-CoA, Glycin und Eisen entstehen mit Hilfe von acht Enzymen Häm-Moleküle [1]. Porphyrien sind Störungen der Häm-Synthese. Sie werden nach der Hauptlokalisation der Synthesestörung als hepatische versus erythropoetische Porphyrien, klinisch als akute versus nicht akute Porphyrien (Abb. 1) definiert. Bei akuten hepatischen Porphyrien (AHP; Kasuistik 1) – akut intermittierende Porphyrie (AIP), Porphyria variegata (PV; Kasuistik 3), hereditäre Koproporphyrie (HCP) und seltene Doss-Porphyrie (ALAS-Defizienz-Porphyrie) – führt die Akkumulation wahrscheinlich neurotoxischer Metaboliten – δ-Aminolävulinsäure (ALA) und Porphobilinogen (PBG) – zu abdominalen, neurologischen, psychiatrischen und kardiovaskulären Symptomen. Dagegen leiden Patient:innen mit nicht akuten Porphyrien – Porphyria cutanea tarda (PCT), erythropoetische Protoporphyrie (EPP; Kasuistik 2), X-linked Protoporphyrie (XLP) und kongenitale erythropoetische Porphyrie (CEP) – infolge stromaufwärts akkumulierter Metaboliten der Häm-Synthese unter Photodermatosen und Leberschäden [1].
Die Regulation der hepatischen Hämsynthese erfolgt durch das geschwindigkeitsbestimmende Enzym δ-Aminolävulinsäuresynthase 1 (ALAS1). ALAS1 wird durch bestimmte Medikamente, Xenobiotika, Alkohol, Fasten, Stress und Inflammation induziert und das Endprodukt Häm inhibiert. Die erythropoetische Hämsynthese wird über ALAS2 mittels eisenregulierenden Transkriptionsfaktoren gesteuert [3].
Der Weg bis zur Diagnose einer Porphyrie kann Jahre dauern und für Betroffene qualvoll sein. Entscheidend ist deshalb, Porphyrien in das diagnostische Kalkül einzubeziehen. Anhand des Musters von Porphyrinvorläufern und Porphyrinen in Urin, Stuhl und Blut lassen sich Porphyrien sicher ausschließen oder diagnostizieren.
Akute hepatische Porphyrien (AHP)
In Deutschland ist die AIP die häufigste akute hepatische Porphyrie (Prävalenz 0,6/100.000) [14].
Die reduzierte Enzymaktivität von circa 50 % der Porphobilinogen-Desaminase (PBGD) ist unter physiologischen Bedingungen ausreichend, um die Hämsynthese zu kompensieren. Die Induktion der ALAS1 (Medikamente, weibliche Sexualhormone, Inflammation, Fasten, Rauchen, Stress und Alkoholabusus) führt zur Dekompensation des defekten Syntheseschritts und zur Akkumulation wahrscheinlich neurotoxischer Metaboliten (ALA und PBG, bei der Doss-Porphyrie und der Bleivergiftung nur ALA) [1]. Die Schädigung des autonomen und peripheren Nervensystems manifestiert sich in abdominellen, neurologischen, kardiovaskulären und psychiatrischen Symptomen (Schmerzen des Abdomens, der Extremitäten und des Rückens, Parästhesien und Lähmungen, Müdigkeit, Übelkeit, Erbrechen, Obstipation, Halluzinationen, Hypertonie). Durch ein Syndrom der inadäquaten ADH-Sekretion (SIADH) kommt es zur Hyponatriämie. Zusätzlich leiden die Patient:innen (mitunter täglich) unter chronischen Beschwerden wie Übelkeit, Antriebslosigkeit (Fatigue), Schmerzen und Angst [7].
Bei PV, HCP und AIP mit schwerer Niereninsuffizienz können zudem Erosionen und Blasen an sonnenlichtexponierten Arealen auftreten [14].
Diagnostik und Therapie
Bei V. a. eine akute hepatische Porphyrie genügt primär die kreatininbezogene Bestimmung von ALA, PBG und den Gesamtporphyrinen im Spontanurin (20 ml). Ein Lichtschutz ist bei zügigem Versand in ein Labor nicht erforderlich. Bei symptomatischen Patient:innen kann die Diagnose einer AHP bei mehr als vierfacher Erhöhung von PBG und ALA gestellt werden [12]. Bei solitärer Erhöhung von ALA muss man eine Doss-Porphyrie, eine Bleiintoxikation oder eine Tyrosinämie Typ 1 in Betracht ziehen. Bei positivem Befund sollte man später zur Differenzierung der akuten Porphyrien die Enzymaktivität der PBGD und die Stuhlporphyrine bestimmen. Im Verlauf sind Mutationsanalysen sowie eine genetische Beratung und die Untersuchung von Familienangehörigen sinnvoll.
Die Basistherapie einer AHP besteht in der ausführlichen Analyse und der Aufklärung über potenziell schubauslösende Trigger. Patient:innen sollten porphyrinogene Medikamente (u. a. Metamizol, Barbiturate), Xenobiotika, Alkohol, Rauchen, Stress und Hormone, die zur Induktion der ALAS1 führen, vermeiden [13]. Medikamente lassen sich auf http://drugs-porphyria.org schnell und sicher auf ihre Porphyrinogenität überprüfen. Eine katabole Stoffwechsellage, z.B. durch Fasten, kann einen Schub hervorrufen [5]. Akut sind eine ausreichende Kalorienzufuhr (Glukose) und eine symptomatische Therapie mit verträglichen Medikamenten notwendig. Bei neurologischer Symptomatik und Hyponatriämie besteht die Indikation zur Therapie mit Hämarginat [14].
Die wiederholte, teils prophylaktische Anwendung von Hämarginat muss aufgrund von Tachyphylaxie, Eisenüberladung und Vasotoxizität kritisch betrachtet werden [15]. Das RNA-Interferenz-Medikament Givosiran ist seit März 2020 in Deutschland bei akuter hepatischer Porphyrie zugelassen. Givosiran (Givlaari®) hemmt selektiv die mRNA der hepatischen, hochregulierten ALAS1, die so wieder normalisiert wird. ALA und PBG fallen deutlich ab. Korrespondierend kommt es zu einer signifikanten Reduktion der Schübe, der Beschwerden und des Hämarginatbedarfs.
Erythropoetische Protoporphyrie (EPP)
Bei der häufigsten Protoporphyrie, der erythropoetischen Protoporphyrie (Prävalenz 0,9/100.000), führt die Einschränkung des letzten Enzyms der Hämsynthese im Knochenmark, der Ferrochelatase, zur Anhäufung von metallfreiem Protoporphyrin IX in den Erythrozyten. Akkumuliertes Protoporphyrin IX hat an lichtexponierten Hautarealen Brennen, Jucken, Schmerzen, Erytheme und Ödeme zur Folge. Protoporphyrin IX wird biliär eliminiert. Bei hepatischer Akkumulation kann es zur Leberschädigung bis zur Leberzirrhose kommen [2].
Diagnostik und Therapie
Die Diagnose der EPP wird durch erhöhte Konzentration von metallfreiem Protoporphyrin IX im antikoagulierten Vollblut (Erythrozyten und Plasma) gestellt [6].
Als therapeutischer Standard galten bislang der Lichtschutz (Hut, Handschuhe, Schirm) und der Leberschutz (Ursodesoxycholsäure, Colestyramin, Vitamin D, Hepatitis-A- und -B-Impfschutz). In schweren Fällen kann man als Ultima Ratio eine allogene Stammzelltransplantation – gegebenenfalls in Kombination mit einer Leber-Tx – erwägen [10, 16]. Seit 2017 ist das α-MSH-Analogon Afamelanotid (Scenesse®) in Deutschland verfügbar. Das s.c. implantierte Afamelanotid wirkt antientzündlich und induziert als selektiv wirkendes Melanocortin-1-Rezeptor-Analogon die Produktion des schwarz-braunen Pigments Eumelanin. Afamelanotid führt bei Patient:innen mit EPP zu einer signifikant besseren Lichttoleranz und Lebensqualität [9]. Es gibt Hinweise, dass unter dieser Medikation die Protoporphyrinkonzentration im Blut und somit die hepatische Schädigung abnehmen.
Porphyria cutanea tarda (PCT)
Die PCT ist die häufigste nicht akute hepatische Porphyrie (Prävalenz 2/100.000). Sie wird durch eine erworbene oder hereditäre Aktivitätsminderung der hepatischen Uroporphyrinogen-Decarboxylase verursacht. Meist führen zusätzliche Trigger zum Ausbruch. Der wichtigste Prädispositionsfaktor ist Eisen, das Uroporphyrinogen zu Porphomethen oxidiert, einem Hemmstoff der Uroporphyrinogen-Decarboxylase [11]. So überrascht es nicht, dass man Hämochromatose HFE-Gen-Mutationen (meist heterozygot) bei etwa zwei Dritteln der Patient:innen findet. Zudem kann die PCT durch Faktoren wie Hexachlorbenzol, Dioxin, Alkohol, hormonale Kontrazeptiva/postmenopausale Hormonersatztherapie, Hämodialyse, HIV und Hepatitis C ausgelöst werden. Die PCT zeigt sich mit Blasen, Narben und erhöhter Verletzlichkeit der sonnenlichtexponierten Hautpartien sowie mit einer Hypertrichose auf Wangen, Schläfen und Jochbein. Neben den erst später auftretenden kutanen Veränderungen kommt es schon früh zur Porphyrinbeladung der Leber. Ähnlich dem Bild einer NAFLD zeigen sich erhöhte Leberenzyme (ALT > AST; ALT: Alanin-Aminotransferase, AST: Aspartat-Aminotransferase). Für die PCT können sich sonografisch fokal echoreiche Strukturen ("Olympiaringe") im Leberparenchym (Abb. 2) zeigen.
Diagnostik und Therapie
Bei der PCT sind die Porphyrine mit einer Dominanz von Uroporphyrin im Serum und Urin erhöht, ALA und PBG im Urin normal. Das Isokoproporphyrin im Stuhl ist erhöht. Im Leberbiopsat lassen sich rot fluoreszierende Porphyrine unter UV-Licht darstellen (Abb. 3) [14]. Die PCT-Therapie basiert auf konsequenter Vermeidung von Auslösern (Alkohol, Hormoneinnahme, Hepatitis-C-Eradikation [4], HIV-Therapie), der Eisendepletion via Aderlass bei Eisenüberladung/HFE-Mutation und der Porphyrinelimination durch "Low dose"-Hydroxychloroquin (HCQ; 2 x 200 mg pro Woche) [8].
Sekundäre Porphyrinurien
Verschiedene Störungen und Medikamente können zu asymptomatischen Porphyrinerhöhungen führen. Dabei sind vor allem Koproporphyrin (Urin, Serum) und Zink-Protoporphyrin im Blut erhöht. Im Zweifel sollte anhand einer kompletten Porphyrinanalytik (Blut, Stuhl und Urin) gemeinsam mit einem Porphyriezentrum eine genaue Einschätzung erfolgen. Blei hemmt das zweite Enzym der Hämsynthese (ALA-Dehydratase). Die Bleiintoxikation führt zu akuten porphyrieähnlichen Symptomen und einer mehr als zehnfachen Erhöhung der ALA im Urin bei normalem oder nur mäßig erhöhtem PBG. Erhöhte Bleiwerte im Urin und Blut bestätigen die Diagnose. In den Erythrozyten kommt es zur "basophilen Tüpfelung".
Fazit
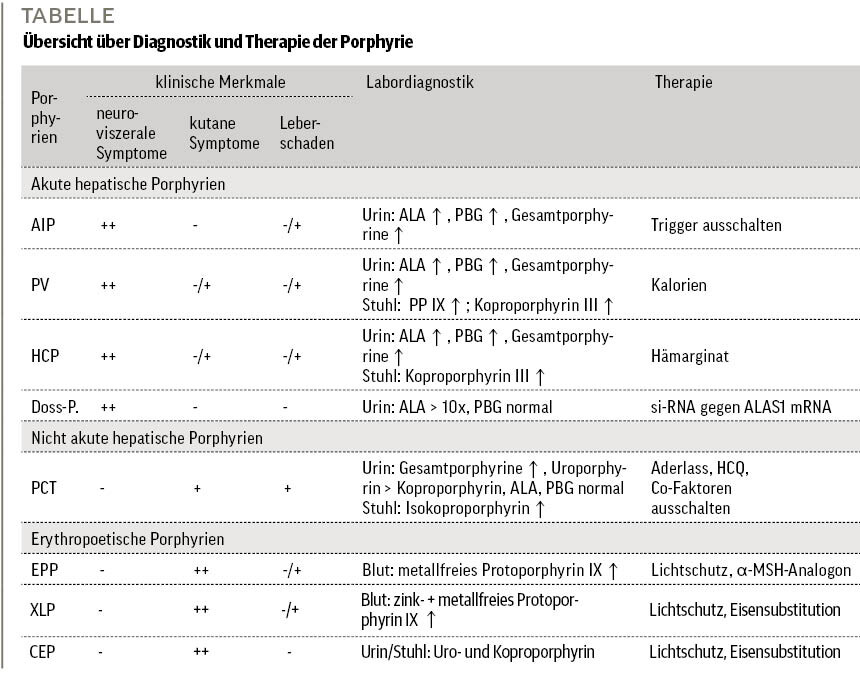
Denken Sie an Porphyrien! Mit ihrem klinischen Bild (neuroviszerale vs. kutane Symptome) und durch Urin-, Stuhl- und Blutproben gelingt es immer, sie zu diagnostizieren (Tabelle). Diagnostik und Therapie sollten abgestimmt mit einem Porphyriezentrum erfolgen. Eine frühe Diagnose verhindert Folgeschäden – vor allem bei akuten hepatischen, erythropoetischen Porphyrien einen prolongierten, traumatisierenden Krankheitsverlauf. Durch neue, gezielte Medikamente lässt sich dieser nachweislich positiv beeinflussen. Mit Givosiran bei akuten Porphyrien und Afamelanotid bei Protoporphyrien sind hocheffektive Therapieoptionen verfügbar.

Dr. med. Ilja Kubisch
Interessenkonflikte: Der Autor hat Vortragshonorare von Alnylam erhalten.
Erschienen in: doctors|today, 2021; 1 (2) Seite 40-43