





Die Haut ist eines der am häufigsten von unerwünschten Arzneimittelwirkungen (UAW) betroffenen Organe. Nicht-lebensbedrohliche Reaktionen wie das makulopapulöse Exanthem, das fixe Arzneimittelexanthem oder die Urtikaria haben dabei die höchste Inzidenz. Seltener sind schwere lebensbedrohliche Arzneimittelwirkungen, die der Hausarzt jedoch trotzdem kennen sollte, weil sie ein schnelles Handeln erfordern.
Prinzipiell kann jedes Medikament eine kutane unerwünschte Arzneimittelwirkung (kUAW) in unterschiedlicher klinischer Manifestation auslösen. Ca. 15 % der UAW beruhen auf allergischen Reaktionen [1]. Antibiotika sind die häufigsten Auslöser für makulopapulöse Exantheme. Schwere kUAW wie Stevens-Johnson-Syndrom (SJS) oder toxisch-epidermale Nekrolyse (TEN) werden dagegen am häufigsten durch Sulfonamide, Allopurinol, gewisse Antikonvulsiva, Nevirapin und Oxicam-Analgetika hervorgerufen. Eine akute generalisierte exanthematische Pustulose (AGEP) wird am häufigsten durch Antibiotika wie Pristinamycin und Aminopenicilline, gefolgt von Chinolonen und Sulfonamiden ausgelöst. Ein Hypersensitivitätssyndrom (DRESS-Syndrom) wird besonders häufig durch bestimmte Antiepileptika und Allopurinol sowie andere Medikamente wie z. B. Minocyclin [2] hervorgerufen.
Ätiologisch sind nicht-immunvermittelte UAW (Typ-A-Reaktionen, 85 - 90 % der Fälle) deutlich häufiger als immunvermittelte UAW (Typ-B-Reaktionen). Andere immunvermittelte Erkrankungen wie Psoriasis, Vaskulitiden und blasenbildende Erkrankungen (Pemphigus, bullöses Pemphigoid) können ebenfalls selten durch Medikamente ausgelöst werden. Klinische Warnsignale für einen schweren und ggf. lebensbedrohlichen Verlauf sind Schleimhautbeteiligung, Blasenbildung und systemische Symptome wie Fieber oder starkes Krankheitsgefühl.
Makulopapulöses Exanthem
Exanthematische Reaktionen treten i. d. R. Tage bis Wochen nach Einnahme eines Medikamentes auf und sind durch eine variable Morphologie und Lokalisation gekennzeichnet. Es findet sich ein generalisiertes, stamm- und extremitätenbetontes, juckendes, unterschiedlich dichtes Exanthem (Abb. 1), meist unter Aussparung des Gesichtes mit Primäreffloreszenzen wie Maculae, Papeln, selten limitierte Pusteln, Bläschen oder Blasen, gefolgt von Sekundäreffloreszenzen wie Schuppen, selten Erosionen oder Hämorrhagien. Differentialdiagnostisch muss eine infektiöse, am häufigsten virale Genese (AZ-Verschlechterung, Fieber, Beteiligung der Schleimhäute, CRP-Erhöhung, Rhinitis, Pharyngitis, Bronchitis, Lymphknotenschwellungen) in Betracht gezogen werden. Zu den häufigsten Auslösern zählen Antibiotika, Tuberkulostatika, Antikonvulsiva und Antihypertensiva.
Therapie: Absetzen des auslösenden Medikamentes, Antihistaminika, abhängig von der Klinik systemische Glukokortikoide (initial Prednisolon 50 - 150 mg/Tag p.o./i.v., im weiteren Verlauf krankheitsadaptiert reduzieren) und extern ggf. milde topische Glukokortikoide (Hydrokortison-Creme 1 %), rückfettende Externa bzw. juckreizstillende Externa (z. B. Optiderm® Creme). Nach sechs Wochen bis maximal sechs Monaten sollten die Hautveränderungen komplett abgeheilt sein [3].
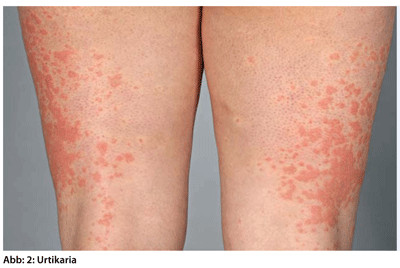
Urtikaria
Eine lokalisierte oder generalisierte, akut, innerhalb von Minuten aufgetretene Urtikaria zeichnet sich durch erhabene, palpable, scharf begrenzte, stark juckende, solitäre bis konfluierende weißlich bis rote Quaddeln unterschiedlicher Größe aus (Abb. 2). Ein in 50 % der Fälle assoziiertes Angioödem stellt eine klinische Sonderform mit Schwellung des Gesichtes, besonders der Lippen und Augenlider, dar. Auslöser können Medikamente, Nahrungsmittel und Infektionen sein. Die Urticae heilen i. d. R. innerhalb von ein bis zwei Wochen ab. Zum Vorgehen in Notfallsituationen vgl. Kasten.
Therapie: Bei arzneimittelassoziierter Urtikaria Absetzen des auslösenden Agens, Antihistaminika p.o. (generalisierte Form: intravenös), abhängig von der Klinik systemische Glukokortikoide (initial Prednisolon 50 - 150 mg/Tag p.o./i.v., im weiteren Verlauf krankheitsadaptiert reduzieren) und extern ggf. milde topische Glukokortikoide (Hydrocortison-Creme 1 %), kühlende feuchte Umschläge im Gesicht (NaCl-Lösung) bzw. juckreizstillende Externa (z. B. mit Menthol oder Polidocanol, s. o.). Topische Antihistaminika zeigen nur mäßige Erfolge und bergen die Gefahr der Kontaktsensibilisierung!
Akute generalisierte exanthematische Pustulose (AGEP)
Eine AGEP ist eine akute und schwere Hautreaktion und zeichnet sich durch stecknadelkopfgroße, nicht den Haarschäften zugeordnete, sterile Pusteln auf einem ödematösen Erythem aus, begleitet von Fieber und Leukozytose (Abb. 3). Die häufigsten Auslöser sind Medikamente wie Ampicillin/Amoxicillin, Chinolone, Pristinamycin, Sulfonamide, Terbinafin, (Hydroxy-)Chloroquin und Diltiazem und Medikamente aus Gruppen wie Antimykotika, Analgetika, Antihypertensiva und Antikonvulsiva.
Vorausgehende Infektionen können als Trigger dienen. Differentialdiagnostisch kommt vor allem eine Psoriasis pustulosa generalisata in Frage.
Therapie: Absetzen des auslösenden Agens, Fokussuche, bei Fokus: adäquate Therapie, Antihistaminika p.o. (ggf. systemisch), abhängig von der Klinik systemische Glukokortikoide (initial Prednisolon bis 250 mg/Tag i.v., im weiteren Verlauf krankheitsadaptiert reduzieren) und extern austrocknende Maßnahmen mit Lotio alba (ggf. mit Clioquinol), ggf. topische Glukokortikoide (Betamethasonvalerat, Triamcinolonacetonid). Abheilung nach zehn Tagen bis vier Wochen [4].
Hypersensitivitätssyndrom
Das Hypersensitivitätssyndrom oder auch DRESS-Syndrom (drug reaction with eosinophilia and systemic symptoms) beschreibt eine lebensbedrohliche Multiorganreaktion und ist charakterisiert durch ein generalisiertes, makulo-papulöses, lichenoides oder multiformes Exanthem bis hin zur Erythrodermie (Abb. 4), verbunden mit einer erheblichen Störung des Allgemeinzustandes, hohem Fieber, Hepatopathie, Nephropathie, pulmonaler Beteiligung und Lymphadenopathie. Im Labor findet sich eine Leukozytose (> 11 x 109/l) mit atypischen Lymphozyten (> 5 %) und Eosinophilie (bis >1,5 x 109/l). Ein DRESS-Syndrom zeichnet sich durch Besonderheiten wie ein verzögertes Auftreten (zwei bis sechs Wochen), eine Verschlechterung der Symptome nach Absetzen des auslösenden Agens und eine unerklärbare Kreuzreaktivität zu nicht-strukturverwandten Medikamenten aus. Neben immunologischen Reaktionen spielt gerade bei der Organbeteiligung die Reaktivierung von Epstein-Barr- und humanen Herpes-Viren eine Rolle. Die Mortalität liegt bei ca. 10 %. Als auslösende Medikamente sind Antikonvulsiva (Carbamazepin, Phenytoin, Phenobarbital, Zonisamid), Mexiletin, Lamotrigin, Nevirapin, Allopurinol, Minocyclin, Abacavir, Lopinavir, Dapson, Sulfasalazin, Sulfonamide beschrieben.
Therapie der Wahl sind in schweren Fällen systemische Glukokortikoide (40 - 50 mg/d mit langsamer Reduktion über sechs bis acht Wochen, um ein erneutes Aufflammen zu verhindern). Bei milden Verläufen ohne Reaktivierung von Viren kann mit einem spontanen Abheilen innerhalb von drei Wochen gerechnet werden [7 - 11].
Stevens-Johnson-Syndrom (SJS) und toxisch epidermale Nekrolyse (TEN)
SJS und TEN sind seltene, aber schwere kUAW und stellen einen medizinischen Notfall dar. Die Mortalität liegt beim SJS bei 1 - 5 % und steigt bei der TEN auf 25 - 35 % an, je nach Ausmaß der betroffenen Körperoberfläche, Alter und Komorbiditäten. TEN und SJS kennzeichnen sich durch mehr oder weniger extensive, schmerzhafte, erythematöse und erosive Ablösung von Haut, Konjunktiven und Schleimhäuten (Abb. 5). Eine Ablösung der Körperoberfläche von 10 % ist definiert als SJS, 10 - 30 % als SJS/TEN-Overlap und > 30 % als TEN [12, 13]. TEN und SJS werden als die beiden Enden eines Spektrums von schweren epidermolytischen kUAW aufgefasst, die sich nur in dem Ausmaß der Hautbeteiligung unterscheiden. Auslösende Medikamente sind in den meisten Fällen Allopurinol, Antibiotika, Antikonvulsiva (bes. Carbamazepin), Sulfonamide, Nevirapine und Oxicam-Analgetika.
Aufgrund der hohen Mortalität ist eine schnelle Diagnose und das Absetzen des auslösenden Agens von immenser Wichtigkeit. Die weitere Behandlung sollte auf einer (Verbrennungs-)Intensivstation erfolgen und u. a. eine immunmodulatorische Therapie mit hochdosierten intravenösen Immunglobulinen (3 g/kg KG über drei bis vier Tage) umfassen [14 - 16].
Fazit
Kutane UAW erfordern in ca. 33 % der Fälle eine Hospitalisation der Patienten und stellen eine signifikante Belastung des Gesundheitssystems dar. Durch neue Erkenntnisse in der Diagnostik von Risikofaktoren und Prädispositionen bei schweren, lebensbedrohlichen kUAW könnten diese verhindert und das Outcome von Patienten wesentlich verbessert werden. Patienten mit kutanen oder systemischen UAW sollten immer einen Allergiepass mit sich führen.

Erschienen in: Der Allgemeinarzt, 2012; 34 (16) Seite 44-48