


Eine Eosinophilie kommt häufig vor und ist eine Begleiterscheinung einer Vielzahl von Erkrankungen. Oft ist es der Hausarzt, der eine Vermehrung der eosinophilen Granulozyten in der Routineuntersuchung mehr oder weniger zufällig entdeckt. Seine Aufgabe besteht dann in der Einleitung einer weiterführenden Diagnostik bzw. je nach vorherrschenden Symptomen in der Überweisung an einen entsprechenden Spezialisten.
Eine Eosinophilie von 500/µl oder gar von 1.500/µl im peripheren Blut (Abb. 1) sollte nicht als unbedeutend abgehakt werden, sondern bedarf, falls nicht anderweitig erklärbar, einer diagnostischen Abklärung. Dieses ist besonders deshalb relevant, weil die eosinophilen Granulozyten aus dem Blut in diverse Organe, vor allem in Lymphknoten, Herz, Haut und Gastrointestinaltrakt, wandern können und dort möglicherweise Organschäden hervorrufen.
Klassifikation
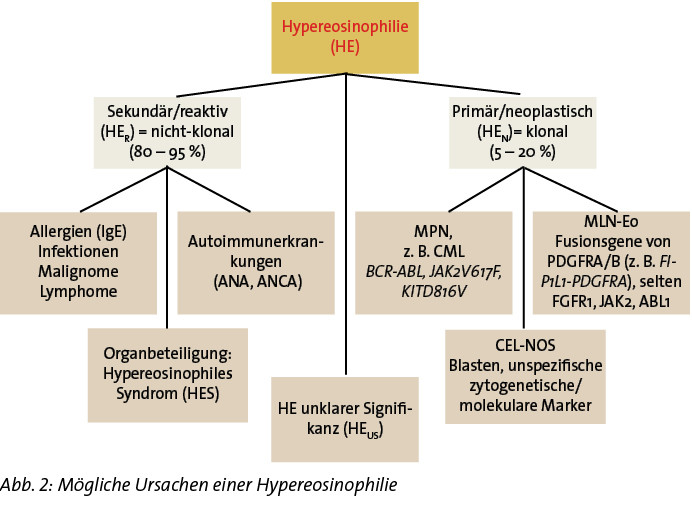
Eine Hypereosinophilie (HE) ist definiert als eine persistierende Eosinophilie im peripheren Blut von mehr als 1.500/µl, bestätigt in zwei Messungen im Abstand von mindestens vier Wochen. Im Jahr 2012 wurden die Kriterien modifiziert und unterschiedliche Subtypen der HE definiert [7] (Tabelle 1).
Die mit ca. 80 % größte Gruppe der HE sind die reaktiven oder sekundären HE (HER), diese entstehen als Begleitphänomen bei Autoimmunerkrankungen, Atopien, Allergien, Infektionen oder Malignomen (Karzinome als auch Lymphome). Ursächlich dafür ist eine Überproduktion von eosinophilopoetischen Zytokinen wie Interleukin-3, Interleukin-5 oder GM-CSF, die die Proliferation von Eosinophilen anregen. Eine HER kann aber auch nach Einnahme von Medikamenten (z. B. Betablocker, ASS) auftreten.
Bei den neoplastischen oder primären HE (HEN) entstammen die Eosinophilien aus dem Stammzellkompartment des Knochenmarks und sind Teil einer klonalen Erkrankung. Hier sind insbesondere myeloproliferative Neoplasien (MPN) zu nennen, wie die chronische myeloische Leukämie (CML), die regelhaft mit einer Eosinophilie einhergeht, myelodysplastische/myeloproliferative Überlappungssyndrome oder seltener akute Leukämien.
Zudem gibt es innerhalb dieser Gruppe die sogenannten myeloischen und lymphatischen Neoplasien mit Eosinophilie und Rearrangierung von PDGFRA, PDGFRB, FGFR1 oder PCM1-JAK2 (MLN-Eo). Die durch Fusion mit verschiedenen Partnergenen entstehenden Fusionsgene (z. B. ETV6-PDGFRB, FIP1L1-PDGFRA) kodieren für eine Tyrosinkinase, deren konsekutive Aktivierung zur Zellproliferation führt.
Die chronische Eosinophilenleukämie (CEL) kann dann diagnostiziert werden, wenn Blasten im peripheren Blut und/oder Knochenmark vorliegen und eine genetische Veränderung (nicht aus der Gruppe der MLN-Eo) vorliegt, die eine Klonalität der Erkrankung nachweist.
Kommt es bei der HE zu einer Eosinophilie-bedingten Organinfiltration oder Organdysfunktion, liegt ein Hypereosinophiles Syndrom (HES) vor [1]. Dieses kann sowohl bei den reaktiven/sekundären oder bei primären/neoplastischen HE auftreten (HESR, HESN).
Bei einer HE, bei der auch nach einer ausgiebigen Ursachenforschung die Genese unklar bleibt und bei der eine Organbeteiligung gezielt ausgeschlossen wurde, wird die vorläufige Diagnose einer HE unklarer Signifikanz (HEUS) gestellt. Diese ist wie bei der monoklonalen Gammopathie unklarer Signifikanz (MGUS) ein kontrollbedürftiger Befund, da nicht bekannt ist, wohin die Reise geht und sich im Verlauf ggf. die bisher nicht diagnostizierte zugrundeliegende Grunderkrankung demaskieren kann.
Symptomatik
Das klinische Erscheinungsbild von Patienten mit HE ist sehr heterogen und abhängig von der Genese sowie dem Vorhandensein bzw. Fehlen einer Organbeteiligung mit entsprechender konsekutiver Funktionsstörung. Es gibt asymptomatische Formen, bei denen die Eosinophilie zufällig bei einer Routineblutabnahme diagnostiziert wird oder blande Krankheitsverläufe (z. B. Beteiligung der Haut mit Pruritus, Urtikaria). Daneben gibt es schwere, potenziell lebensbedrohliche Verläufe mit Organinfiltration und -dysfunktion (z. B. Gastrointestinaltrakt mit Gastritis, Kolitis, Hepatopathie oder Aszites). Insbesondere eine Beteiligung von Lunge (z. B. Asthma bronchiale, Pleuraerguss, pulmonale Infiltrate, Lungenfibrose) und Herz (z. B. intrakardiale Thrombosen, restriktive Kardiomyopathie, Myo- oder Perikarditis, Perikarderguss) sind gefürchtete Komplikationen der HE.
Die MPN-Eo können, wie auch andere MPN (z. B. CML), einen indolenten chronischen Verlauf nehmen mit konstitutionellen Symptomen (Fatigue, Nachtschweiß, Gewichtsverlust), Splenomegalie und Leukozytose bei hyperzellulärem Knochenmark, oder aber als akute aggressive Erkrankung im Sinne einer Blastenphase imponieren. Hierbei entspricht das klinische Bild dem einer akuten myeloischen Leukämie oder dem eines aggressiven Lymphoms.
Insbesondere beim Nachweis eines Asthma bronchiale, einer Sinusitis mit einer bioptisch gesicherten Vaskulitis und ggf. Nachweis anti-neutrophiler cytoplasmatischer Antikörper und renaler Beteiligung muss differenzialdiagnostisch an eine eosinophile Granulomatose mit Polyangiitis (EGPA), früheres Churg-Strauss-Syndrom (CSS), gedacht werden.
Diagnostik
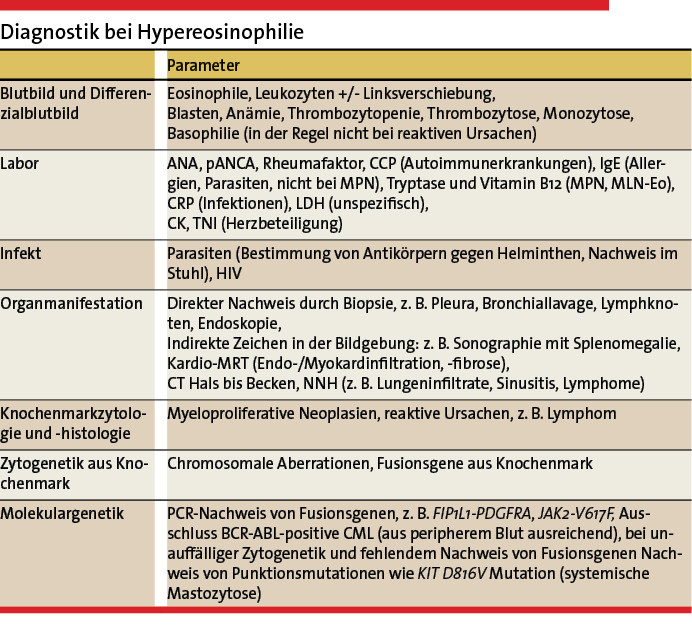
Das vorrangige Ziel einer HE-Diagnostik sollte – aus prognostischer und therapeutischer Sicht – zunächst die eindeutige Unterscheidung zwischen nicht-klonaler HER und klonaler HEN sein. Die Säulen der Diagnostik bilden die Befunde aus Blutbild, Serumchemie, bildgebenden Verfahren inklusive Endoskopie, Organhistologie und vor allem aus molekularzytogenetischer Untersuchung von Blut und Knochenmark (Tabelle 2, Abb. 2) [6].
Die Blutbildveränderungen, insbesondere die absolute Anzahl der Eosinophilen im peripheren Blut, sind leider nicht richtungsweisend und können für die Typisierung des HE nicht herangezogen werden. Begleitphänomene wie eine Leukozytose > 50/nl, eine Thrombozytopenie < 100/nl oder auch eine Thrombozytose > 1 Mio/nl, sowie eine pathologische Linksverschiebung bis hin zu Blasten sind Veränderungen, die mit einem reaktiven Geschehen in der Regel nicht mehr vereinbar sind, hier sollte zeitnah eine weiterführende Abklärung, vor allem zum Ausschluss einer zugrundeliegenden MPN, erfolgen. Ein erhöhter Vitamin-B12-Spiegel im Serum (ohne vorherige Substitution) ist ebenfalls typisch (aber unspezifisch) für eine neoplastische Genese (u. a. CML, MLN-Eo).
Bei einem kleinen Anteil der Patienten mit HEN kann eine erhöhte Serumtryptase diagnostisch wegweisend sein (u.a. MLN-Eo). Bei einer Serumtryptase von > 20/µl sollte insbesondere eine systemische Mastozytose molekulargenetisch (KITD816V-Mutation) und durch eine Knochenmarkpunktion mit Histologie ausgeschlossen werden. Bei etwa 20 – 30 % der systemischen Mastozytosen zeigt sich im peripheren Blutbild eine Eosinophilie. Auch eine erhöhte alkalische Phosphatase (AP), Aszites und Splenomegalie sind klinische Zeichen einer in der Regel fortgeschrittenen systemischen Mastozytose.
Praktisches Vorgehen
Es gibt mehrere Ansätze, wie die Ursache einer Eosinophilie abgeklärt werden kann. Zunächst sollte man möglichst alle reaktiven Ursachen der Reihe nach ausschließen. Dies bedeutet zunächst die Bestimmung von Entzündungswerten wie CRP, IgE, Autoantikörpern (u.a. ANA, ANCA, CCP). Eine IgE-Erhöhung im Serum oder der Nachweis von Rheumafaktoren/citrulliniertem Peptid sind häufiger mit einer reaktiven HE, vor allem mit Allergien und Autoimmunerkrankungen, assoziiert. Bei Nachweis von Autoantikörpern ist eine rheumatologische Abklärung notwendig, um eine eventuell zugrundeliegende Autoimmunerkrankung zu klassifizieren. Hier sind vor allem die eosinophile Granulomatose mit Polyangiitis (EGPA), ehemals Churg-Strauss-Syndrom (CCS), zu nennen. Diagnostisch wegweisend ist eine Biopsie (Nachweis einer Vaskulitis) sowie der Nachweis antineutrophiler cytoplasmatischer Antikörper (p-ANCA, in etwa 40 % nachweisbar). In einem nicht geringen Teil der Fälle passt das Antikörpermuster jedoch nicht komplett zu einer spezifischen Autoimmunerkrankung, die Biopsie ist negativ oder unspezifisch, aber der Patient ist symptomatisch. Hier muss eine autoimmune Genese ggf. angenommen werden und ein Therapieversuch mit Steroiden erfolgen.
Eine parasitäre Erkrankung, insbesondere eine Wurminfektion, ist in unseren Breitengraden eher selten Ursache einer HE, sollte aber insbesondere bei Rückkehrern aus dem Urlaub oder bei Migranten in Erwägung gezogen werden.
Bei einer HE unklarer Genese ist es aus therapeutischer und prognostischer Sicht essenziell, eine Organbeteiligung gezielt nachzuweisen bzw. auszuschließen (vgl. Tabelle 2). Eine Beteiligung des Herzens sollte, aufgrund der potenziell lebensbedrohlichen Komplikation, auch bei asymptomatischen Patienten ausgeschlossen werden. Eine endoskopische Diagnostik mit ÖGD und Koloskopie ist je nach Klinik des Patienten zum Ausschluss eines gastrointestinalen Befalls erforderlich. Weitere Bildgebung richtet sich nach den führenden Symptomen.
Myeloische HE erfordern in der Regel eine Knochenmarkpunktion mit histologischer Aufarbeitung, eine komplette Zytogenetik und eine molekulargenetische Analyse [5]. Die häufigste Form des MLN-Eo ist mit dem FIP1L1-PDGFRA-Fusionsgen assoziiert, das mittels RT-PCR aus dem EDTA-Blut bestimmt werden kann [2].
Therapie
Die Therapie der HE ist abhängig von der Genese, dem Subtyp und der klinischen Symptomatik.
Bei reaktiven HE steht die Behandlung der Grunderkrankung im Vordergrund. Entsprechend den Empfehlungen bei klassischen Autoimmunerkrankungen basiert die Behandlung des HES auf der systemischen Gabe von Steroiden. Hierunter zeigt sich in der Regel ein rascher (innerhalb von ein bis zwei Wochen) Abfall der Eosinophilenzahlen im peripheren Blut. Manchmal sind Steroiddosen von mehr als 10 mg/Tag notwendig, um die Symptome und die Eosinophilie zu kontrollieren. Hier sollte frühzeitig eine Umstellung auf andere steroidsparende Immunsuppressiva in Betracht gezogen werden (u. a. Azathioprin, Cyclophosphamid, Methotrexat).
Patienten mit Nachweis von Fusionsgenen unter Beteiligung von PDGFRA (z. B. FIP1L1-PDFRA) oder PDGFRB (z. B. ETV6-ABL) zeigen selbst in der Blastenphase (klinisches Bild einer Leukämie oder eines lymphoblastischen Lymphoms) ein exzellentes Ansprechen auf eine Monotherapie mit dem Tyrosinkinaseinhibitor Imatinib mit einer dauerhaften kompletten Remission in nahezu allen Fällen [4]. Nicht selten erhalten die Patienten in Unkenntnis des Fusionsgens jedoch für das vermutete "typische" Lymphom oder die Leukämie eine intensive Therapie bis hin zur allogenen Stammzelltransplantation [3]. Dadurch werden, wenn überhaupt, nur vorübergehende kurze Remissionen erreicht. Nahezu alle Patienten rezidivieren. Erst aufgrund der persistierenden Eosinophilie wurde in diesen Fällen nachfolgend das Fusionsgen identifiziert und eine adäquate Therapie mit Imatinib eingeleitet. Daher ist es wichtig, solche Patienten, bei denen es sich ganz überwiegend um Männer handelt, zu identifizieren.
Aufgrund der schlechten Prognose von Patienten mit FGFR-Fusionsgen müssen die Patienten frühzeitig einem Transplantationszentrum vorgestellt werden, da nur eine allogene Stammzelltransplantation potenziell kurativ ist.
Aufgrund der klinischen und genetischen Heterogenität der CEL-NOS existiert bis heute keine standardisierte Behandlungsempfehlung. Hydroxyurea wird zur Kontrolle der Eosinophilie, Leukozytose und Splenomegalie eingesetzt.

Interessenkonflikte: Die Autorin hat keine deklariert.
Erschienen in: Der Allgemeinarzt, 2017; 39 (9) Seite 60-64