



Kommt es bei einem scheinbar gesunden Menschen zur akuten Blutung ist neben fokussierter Anamnese und körperlicher Untersuchung eine zielgerichtete hämostaseologische Diagnostik indiziert. Grundsätzlich ist dabei an Störungen der Thrombozytenfunktion (primäre Hämostase), der plasmatischen Gerinnung (sekundäre Hämostase) oder der Gefäße zu denken. Häufig sind jedoch mehrere Systeme gleichzeitig betroffen.
Was die Ursache einer Gerinnungsstörung angeht, wird zwischen angeborenen und erworbenen Erkrankungen unterschieden (Abb. 1). Letztere können entweder primär das Hämostasesystem betreffen oder sekundär aus einer anderen Grunderkrankung resultieren. Blutungen aus „voller Gesundheit“ sind meist Folge einer erworbenen Gerinnungsstörung.
Angeboren oder erworben?
Bei der Abklärung, ob es sich um eine angeborene oder erworbene Hämostasestörung handelt, sind gezielte Fragen nach Verletzungen oder Operationen in der Vergangenheit sinnvoll. Hierbei sollte insbesondere nach Eingriffen in der Mundhöhle und im Rachenbereich gefragt werden (z. B. Zahnextraktion oder Polyp-/Tonsillektomie). Denn diese Operationen führen bei Patienten mit ansonsten milden hämorrhagischen Diathesen typischerweise zu Blutungskomplikationen. Bei Frauen sind eine verstärkte (Hypermenorrhoe) oder verlängerte Menstruationsblutung (Menorrhagie) sowie die Angabe von peripartalen Blutungskomplikationen zusätzliche Hinweise für eine angeborene Hämostasestörung.
Die Frage nach symptomatischen Familienangehörigen kann die Blutungsneigung weiter eingrenzen. So sind bei der klassischen Bluterkrankheit, der Hämophilie A oder B, die X-chromosomal rezessiv vererbt werden, fast ausnahmslos männliche Familienmitglieder betroffen, während das angeborene Von-Willebrand-Syndrom aufgrund seines autosomalen Erbgangs keine Geschlechterpräferenz zeigt (sog. Pseudohämophilie).
Krebsleiden, Leber- und Niereninsuffizienz sind häufig mit komplexen Störungen der primären und sekundären Hämostase assoziiert. Zudem können endokrinologische Erkrankungen (z. B. Hypothyreose) eine Blutungsneigung fördern [2].
Im Rahmen der Medikamentenanamnese sollte gezielt nicht nur nach der Einnahme von gerinnungshemmenden Medikamenten (z. B. Marcumar®), sondern auch nach Schmerztabletten gefragt werden, da Acetylsalicylsäure und nichtsteroidale Antirheumatika über eine Hemmung der thrombozytären Cyclooxygenase-1 die Plättchenfunktion nachhaltig beeinflussen können. Auch für Antidepressiva aus der Gruppe der selektiven Serotonin-Wiederaufnahmehemmer wurden antiaggregatorische Effekte beschrieben [4].
Wie sieht die Blutung aus?
Lokalisation, Typ und Zeitpunkt einer Blutung können wichtige Informationen über den zugrunde liegenden Hämostasedefekt liefern.
Störungen der primären Hämostase manifestieren sich häufig als flohstichartige (Petechien) oder kleinfleckige Blutungen (Purpura) in abhängigen Körperpartien und als Blutungen im Schleimhautbereich, was zu Epistaxis, Zahnfleischbluten oder Hypermenorrhoe und Menorrhagie führen kann. Typischerweise kommt es unmittelbar nach einer Verletzung oder einem operativen Eingriff zur Blutung.
Störungen der sekundären Hämostase manifestieren sich meist als großflächigere, konfluierende Einblutungen in Weichteilgewebe wie Haut (Sugillationen, Ekchymosen und Suffusionen) oder Skelettmuskulatur sowie, bei schwerer Ausprägung, als spontane Gelenkeinblutung (Hämarthros). Postoperativ kommt es nach anfänglich scheinbar suffizienter Hämostase zur verzögerten Nachblutung.
Labordiagnostik
Bei der analytischen Abklärung einer unklaren Blutungsneigung sollten zunächst die folgenden drei Parameter bestimmt werden, die ohne größeren technischen Aufwand kurzfristig verfügbar sind:
- Thrombozytenzahl
- Aktivierte partielle Thromboplastinzeit (APTT)
- Thromboplastinzeit (Quick-Wert)
Thrombozytenzahl
Thrombozyten sind für eine effektive primäre Hämostase von zentraler Bedeutung. Hierbei spielen sowohl Quantität als auch Qualität eine Rolle. Spontane Blutungen sind, eine regelrechte Funktion vorausgesetzt, erst bei einer Plättchenzahl von < 20 000/µl zu erwarten. Während eine Thrombozytopenie rasch zu diagnostizieren ist, sind zur Beurteilung der Plättchenfunktion aufwendigere Labortests erforderlich (z. B. Aggregometrie).
APTT und Quick-Wert
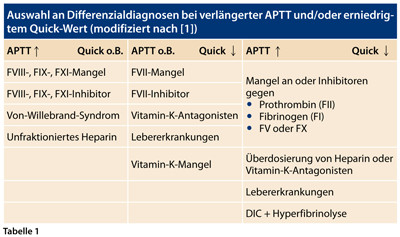
Auch wenn diese Globaltests nur bedingt die in vivo ablaufenden Gerinnungsprozesse widerspiegeln, liefern sie wichtige Informationen über die Ätiopathogenese einer Blutungsneigung. Dabei ist die Kenntnis der in das jeweilige Testsystem einfließenden Einzelfaktoren von Bedeutung (Tabelle 1). Eine korrekte Interpretation der Labordiagnostik ist jedoch nur unter Berücksichtigung der klinischen Beschwerdesymptomatik möglich.
Im Folgenden soll auf ausgewählte Hämostasestörungen, die einer Blutung aus „voller Gesundheit“ zugrunde liegen können, näher eingegangen werden.
Immunthrombozytopenie
Werden bei einem älteren Menschen mit akuten Schleimhaut- und petechialen Hauteinblutungen deutlich verminderte Thrombozytenwerte gemessen, sollte umgehend der periphere Blutausstrich beurteilt werden, um z. B. eine akute Leukämie auszuschließen. Bei morphologisch regelrechter roter und weißer Reihe, fehlenden Allgemeinsymptomen, leerer Medikamentenanamnese und unauffälliger körperlicher Untersuchung (keine Hepatosplenomegalie oder Lymphadenopathie) kann die Verdachtsdiagnose einer Immunthrombozytopenie (ITP) gestellt werden. Bei der ITP ist die Überlebensdauer zirkulierender Thrombozyten durch Autoantikörper dramatisch verkürzt. Für die chronische Verlaufsform der ITP (Morbus Werlhof) sprechen bereits in vergangenen Jahren registrierte und stetig abnehmende Thrombozytenwerte.
Thrombotisch-thrombozytopenische Purpura
Bestehen neben der Thrombozytopenie Fieber und neurologische Ausfälle und werden im Blutausstrich fragmentierte Erythrozyten (Schistozyten) gesehen, muss der Verdacht auf eine thrombotisch-thrombozytopenische Purpura (TTP) geäußert werden (Moschcowitz-Syndrom). Bei dieser akut lebensbedrohlichen hämolytischen thrombotischen Mikroangiopathie kommt es infolge von Autoantikörpern gegen das Enzym ADAMTS13 zu einem verminderten physiologischen Abbau des hochmolekularen Von-Willebrand-Faktors (VWF), der in der Mikrozirkulation verschiedener Organe (z. B. von Gehirn und Niere) eine spontane Plättchenaggregatbildung mit thrombotischer Vasookklusion induziert [3].
Erworbenes Von-Willebrand-Syndrom
Lässt der Blutungstyp eine Störung der primären Hämostase vermuten (z. B. verlängertes Zahnfleischbluten nach einer Parodontosebehandlung), und ist bei normaler Thrombozytenzahl die APTT verlängert, ist differenzialdiagnostisch ein erworbenes Von-Willebrand-Syndrom (VWS) in Erwägung zu ziehen. Der VWF, ein multimeres Glykoprotein, vermittelt die Adhäsion der Thrombozyten an die verletzte Gefäßwand. Daher sind quantitative und/oder qualitative Defekte des VWF mit einer verlängerten Blutungszeit assoziiert. Das erworbene VWS ist sehr wahrscheinlich häufiger als ursprünglich angenommen, denn zahlreiche Grunderkrankungen wurden mit einem beschleunigten Abbau des VWF in Verbindung gebracht (Tabelle 2). Therapeutisch steht die Behandlung der Grunderkrankung im Vordergrund [5].
Chronische DIC mit Hyperfibrinolyse
Die disseminierte intravasale Gerinnung (DIC) ist eine komplexe Gerinnungsstörung, der verschiedene Grunderkrankungen zugrunde liegen. Insbesondere Malignome, die vielfältige aktivierende Einflüsse auf das Hämostasesystem ausüben, sind diesbezüglich in die Differenzialdiagnose einzubeziehen. Bei der chronischen Verlaufsform ist reaktiv auch die Fibrinolyse gesteigert, was entscheidend zur Blutungsneigung beiträgt. Bei der DIC sind typischerweise Thrombozytenzahl, APTT und Quick-Wert pathologisch verändert. Neben einer Hypofibrinogenämie finden sich meist exzessiv erhöhte Fibrinspaltprodukte (D-Dimere). Durch verschiedene Maßnahmen, die auf eine Bremsung der prokoagulatorischen (z. B. durch Heparin) oder der fibrinolytischen Prozesse (z. B. durch Tranexamsäure) sowie auf eine Substitution verbrauchter Gerinnungsfaktoren abzielen (z. B. durch Frischplasma oder Faktorenkonzentrate), kann versucht werden, die dekompensierte Hämostasestörung kurzfristig zu stabilisieren. Entscheidend ist jedoch auch hier die effektive Therapie der Grunderkrankung.

Erschienen in: Der Allgemeinarzt, 2011; 33 (15) Seite 37-40