

Unter dem Begriff Leukämien werden unterschiedliche Gruppen sehr heterogener Krankheitsbilder zusammengefasst. Die Variabilität dieser Erkrankungen spiegelt sich in den äußerst unterschiedlichen morphologischen, immunphänotypischen, zytogenetischen und molekulargenetischen zellulären Eigenschaften wider. Am Beispiel der CLL und AML wird der heutige Standard des diagnostischen Weges dargestellt.
Ein gemeinsames Merkmal aller Leukämien ist die Vermehrung weißer Blutkörperchen durch klonale Stammzelldefekte. Bei einem Großteil der Patienten zeigt sich die zum Teil extreme Steigerung der Leukozyten im peripheren Blutbild. Bei einigen Patienten finden sich die Leukämiezellen aber praktisch nur im Knochenmark, im peripheren Blut dagegen ist die Leukozytenzahl vermindert. Diese Leukämieform wird als „aleukämische Präsentation“ bezeichnet. In seltenen Fällen finden sich die Leukämiezellen bei Erstdiagnose weder im peripheren Blut noch im Knochenmark, sondern infiltrierend in anderen Geweben, beispielsweise testikulär oder intestinal (Beispiel: myeloisches Sarkom).
Eckpfeiler der Diagnostik
Für eine optimale Diagnostik muss eine genügende Anzahl an Leukämiezellen aus dem peripheren Blut, dem Knochenmark und/oder dem befallenen extramedullären Gewebe gewonnen werden. Die heutige moderne Diagnostik umfasst bei allen Leukämien zwingend:
- einerseits Routineanalysen wie die maschinelle quantitative Analytik und die Morphologie sowie
- andererseits spezialisierte Analysen wie Immunphänotypisierung, Molekulargenetik und Zytogenetik.
Als Referenz gilt die im Jahr 2008 publizierte vierte Edition der WHO-Klassifikation hämatopoietischer und lymphatischer Neoplasien [1].
Je nach Reifegrad der leukämischen Zelle werden chronische von akuten Leukämien und je nach Ursprung der leukämischen Zellen lymphatische von myeloischen Leukämien unterschieden. Im Folgenden wird die detaillierte Diagnostik am Beispiel der chronischen lymphatischen Leukämie und der akuten myeloischen Leukämie beschrieben.
Chronische lymphatische Leukämie
Die chronische lymphatische Leukämie (CLL) ist eine klonale Erkrankung der reifen B-Lymphozyten mit vermehrtem Auftreten dieser Zellen im peripheren Blut [2]. Im diagnostischen Prozess gilt es, die CLL klar von anderen lymphoproliferativen Entitäten der B-Zellen mit leukämischem Verlauf zu unterscheiden. Diese sind u. a. die B-Prolymphozytenleukämie, das lymphoplasmozytische Lymphom (Morbus Waldenström), die Haarzellleukämie und das follikuläre Non-Hodgkin-Lymphom. Die Unterscheidung gelingt durch Kombination von Zellzählung und morphologischer Beurteilung des Blutausstriches (sowie ggf. des Knochenmarkes) mit der Immunphänotypisierung der lymphatischen Elemente.
Charakteristika der CLL
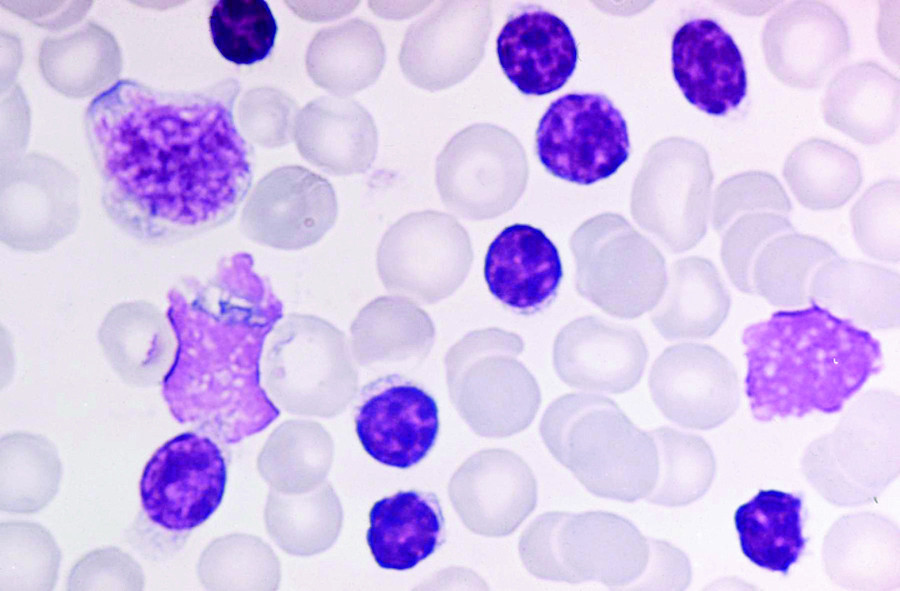
Für die CLL wird eine während mindestens drei Monaten anhaltende Lymphozytose (mehr als 5 G/l) von klonalen B-Lymphozyten verlangt. Morphologisch ist die typische CLL-Zelle klein bis mittelgroß mit schmalem bis fast fehlendem Zytoplasmasaum (nacktkernig) und rundem bis ovoidem Kern mit dichtem Kernchromatin, welches oft charakteristisch marmoriert ist (Abb. 1). Die meist vorkommenden lädierten lymphatischen Zellen im Blutausstrich werden bei Diagnose einer CLL als „Gumprecht‘sche Kernschatten“ bezeichnet. Oft finden sich zusätzlich größere lymphatische Zellen mit gebuchtetem Kern, verfeinertem Kernchromatin und angedeutetem Nukleolus sowie breiterem Zytoplasmasaum. Diese Zellen werden als Prolymphozyten bezeichnet. Je höher der Anteil dieser Zellen ist, desto weniger wahrscheinlich ist die Diagnose einer CLL. Formal darf der Anteil an Prolymphozyten gemäß Hallek [2] bis 55 % aller lymphatischen Elemente ausmachen. In Fällen mit hohem Anteil an Prolymphozyten im peripheren Blut sollte unter Berücksichtigung der Immunphänotypisierung eine Knochenmarkaspiration inklusive Biopsie zur Diagnosesicherung der CLL durchgeführt werden.
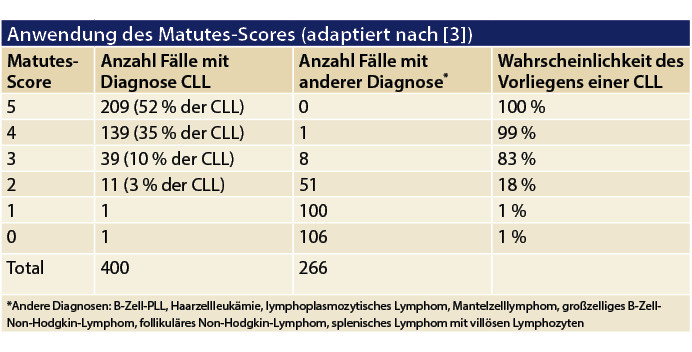
Immunphänotypologisch wird die Klonalität der B-Zellen mittels Darstellung der Oberflächenrestriktion für Kappa- oder Lambda-Leichtketten bestätigt. Die typische CLL zeigt eine deutliche aberrante Oberflächenexpression des T-Zell-Markers CD5 mit deutlicher Expression von CD23 sowie schwacher Positivität für CD79b/CD22 und oberflächlichem Immunglobulin (SIg) bei Negativität für FMC7. Mit diesen fünf Markern kann gemäß Matutes [3]die Wahrscheinlichkeit der Diagnose einer CLL beurteilt werden (Tabelle 1).
Bei eindeutiger Morphologie und passendem Immunphänotyp kann bei einem Patienten ohne Therapieindikation auf eine Knochenmarkpunktion verzichtet werden. Sobald jedoch atypische Verhältnisse in Morphologie und/oder FACS-Analyse vorgefunden werden, sollte zur Diagnosesicherung eine Knochenmarkpunktion inklusive Biopsie und gegebenenfalls Zytogenetik empfohlen werden. Zur prognostischen Einordnung bei Erstdiagnose oder spätestens vor Therapiebeginn werden die klonalen Lymphozyten mittels Fluoreszenz-in-situ-Hybridisierung (FISH) untersucht. Bei rund 80 % der CLL-Patienten finden sich damit genetische Veränderungen, wobei die Deletion des langen Armes von Chromosom 13 (13q-) die häufigste genetische Anomalie darstellt (ca. 50 % der Patienten). Als singuläre Veränderung ist die 13q-Deletion mit einem prognostisch eher günstigen Verlauf assoziiert. Die Deletion des kurzen Armes von Chromosom 17 (17p-) ist dagegen mit einer schlechten Prognose vergesellschaftet, und diese CLL-Patienten sollten nicht mit Fludarabin behandelt werden (hohe Resistenzrate).
Akute myeloische Leukämie
Bei der akuten myeloischen Leukämie (AML) handelt es sich um eine ausgesprochen heterogene Gruppe von Erkrankungen mit sehr unterschiedlichen Prognosen. Die Diagnose erfolgt über die Morphologie des peripheren Blutes und des Knochenmarks in Kombination mit Zytogenetik, Molekulargenetik und Immunphänotypisierung. Diese Untersuchungen erlauben eine Klassifizierung der AML gemäß FAB [4] und WHO 2008 [1, 5] (Tabelle 2).
Beurteilungskriterien
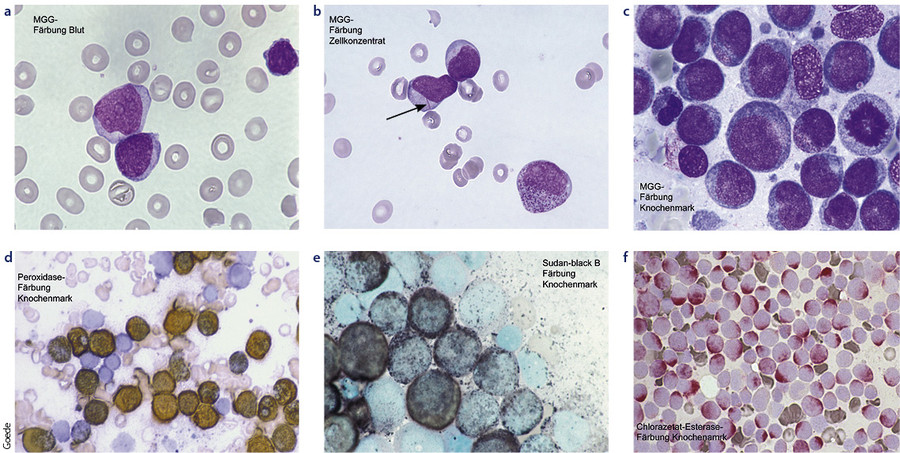
Für die morphologische Beurteilung müssen das periphere Blutbild und eine Knochenmarkaspiration vorliegen. Im peripheren Blut werden mindestens 200 Leukozyten und im Knochenmarkaspirat 500 kernhaltige Zellen mikroskopisch differenziert. Für die Diagnose AML muss der Blastenanteil im Blutbild oder im Knochenmarkaspirat 20 % oder mehr betragen. In der Differenzierung werden Myeloblasten, Monoblasten und auch Megakaryoblasten unter den „Blasten“ differenziert (vgl. Abb. 2).
Zur Beurteilung der Linienzugehörigkeit werden die zytochemischen Spezialfärbungen und die Immunphänotypisierung hinzugezogen. Werden ≥ 3 % Myeloproxidase-positive Blasten ausgezählt, darf die Leukämie der myeloischen Linie zugeordnet werden. Die zytochemische Charakterisierung der Blasten in Kombination mit der noch vorhandenen hämatopoietischen Ausreifung in der May-Grünwald-Giemsa-Färbung erlaubt die Klassifikation der AML gemäß FAB. FAB steht für French-American-British und bezieht sich darauf, dass bei der Ausarbeitung dieser Klassifikation französische, US-amerikanische und britische Experten beteiligt waren.
- FAB M0: AML vom myeloblastären Typ mit minimaler Differenzierung
- FAB M1: AML vom myeloblastären Typ ohne Ausreifung
- FAB M2: AML vom myeloblastären Typ mit Ausreifung
- FAB M3: akute promyelozytäre Leukämie
- FAB M4: AML vom myelomonozytären Mischtyp
- FAB M5a: akute Monoblastenleukämie
- FAB M5b:akute Monozytenleukämie
- FAB M6: akute Erythroleukämie
- FAB M7: akute Megakaryoblastenleukämie
Bei weniger als 3 % Myeloperoxidase-positiven Blasten und bei der akuten Megakaryoblastenleukämie wird die Linienzugehörigkeit mittels Immunphänotypisierung bestimmt respektive bestätigt.
Zytogenetische Analyse der Blasten
Die zytogenetische Analyse der Blasten ist in der Abklärung der akuten Leukämie eine zwingende Untersuchung. In etwas mehr als 50 % der Fälle finden sich dabei Abnormitäten. Um eine AML mit unauffälligem Karyotyp zu diagnostizieren, müssen mindestens 20 Metaphasen aus Knochenmarkaspirat untersucht werden. Bei abnormen Karyotypen können auch die zytogenetischen Ergebnisse aus peripherem Blut berücksichtigt werden.
Mutationsanalysen
Bei jeder AML-Neudiagnose werden routinemäßig genomische DNA und RNA hergestellt sowie Zellen tiefgefroren. Bei limitierter Zellzahl wird der Gewinnung von RNA Vorrang gegeben, weil auf RNA die meisten Fusionsgene und auch Leukämie-assoziierten Mutationen detektiert werden können. In der Molekulargenetik werden folgende Fusionstranskripte gesucht: RUNX1-RUNX1T1, CBFB-MYH11, PML-RARA, MLLT3-MLL und DEK-NUP214. Zusätzlich werden Veränderungen folgender Gene detektiert: NPM1, FLT3, CEBPA, EVI1.
Implikationen für Therapiewahl
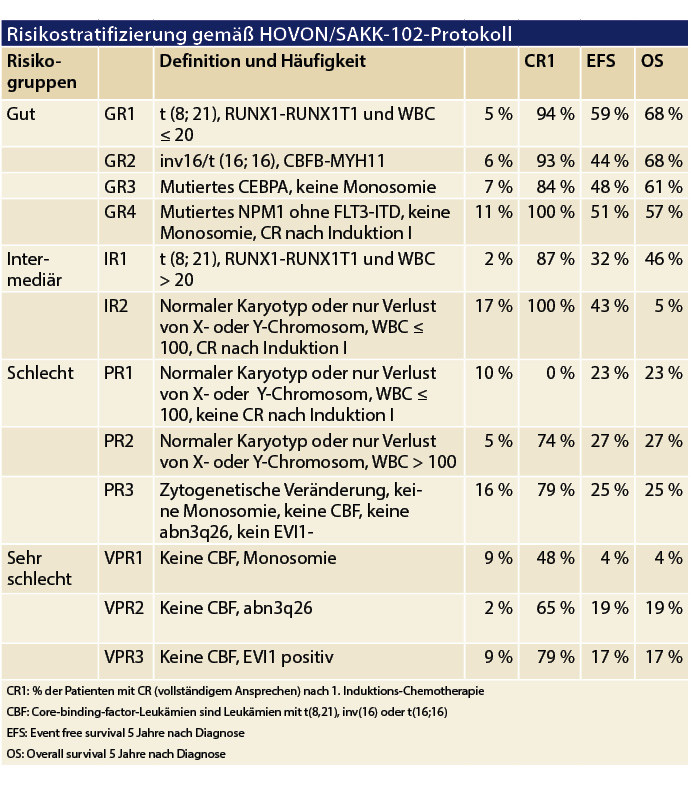
Bei Patienten, die sich für eine intensive Therapie qualifizieren, kann mittels dieser Informationen zusammen mit dem morphologischen Therapieansprechen auf den ersten Induktions-Chemotherapie-Zyklus ein differenziertes Bild bezüglich der Krankheitsprognose erstellt werden. Diese Diagnostik hat unmittelbaren Einfluss auf das therapeutische Vorgehen, indem sie die Indikation einer allogenen Stammzelltransplantation und damit die Notwendigkeit einer Fremdspendersuche bestimmt.
Die Beurteilung des Therapieansprechens nach der ersten Induktions-Chemotherapie kann bei einsetzender Regeneration der Myelopoiese Schwierigkeiten bereiten. Gerade für diese Situationen ist die optimale Charakterisierung der Leukämiezellen bei Erstdiagnose gemäß der klassischen FAB-Klassifikation wichtig. Dies hilft, die einsetzende, noch linksverschoben ausreifende Myelopoiese von leukämischen Blasten abzugrenzen. Deswegen sollte die AML-Diagnostik weiterhin gemäß FAB und WHO 2008 erfolgen.
Durch „gene expression profiling“, kürzlich auch durch "epigenetic profiling" und „next generation sequencing“ ergänzt, konnte das Krankheitsbild der AML auf molekularer Ebene noch wesentlich detaillierter untersucht werden. Dadurch konnte eine Reihe neuer Biomarker festgestellt werden. Ein Teil dieser Marker wird in Zukunft klinische Auswirkungen bei der Beurteilung von Patienten mit AML haben. Zusätzlich haben diese Untersuchungen einmal mehr die Heterogenität der AML mit einer enormen Vielzahl von Mutationen bei Erstdiagnose wie auch im weiteren Krankheitsverlauf aufgezeigt [6]. ▪
Genehmigter und bearbeiteter Nachdruck aus Onkologie 2/2013Ch-

Erschienen in: Der Allgemeinarzt, 2013; (12) Seite 38-42